J Clin Aesthet Dermatol. 2026;19(6):49–51.
by David Seiter, FNP-C, and James Q. Del Rosso, DO
Mr. Seiter is with Epiphany Dermatology, Peoria, Arizona. Dr. Del Rosso is with JDR Dermatology Research, Las Vegas, Nevada, and Touro University Nevada, Henderson, Nevada.
FUNDING: No funding was provided for this article.
DISCLOSURES: The authors have no relevant conflicts of interest.
AUTHOR’S NOTE: The legendary basketball icon Michael Jordan, who led the Chicago Bulls to six NBA championships and was a five-times NBA Most Valuable Player over his 15-year playing career, once said the following: “Get the fundamentals down and the level of everything you do will rise.” As the Editor-in-Chief of the JCAD, author J.D.R. felt that if it was good enough for Michael Jordan, it is good enough for us in dermatology as our goal is always to get smarter and be better. To do this, we have to also maintain and update our core knowledge of fundamentals in dermatology. This is the first article in what is planned to be a series of articles published to assist clinicians in maintaining their knowledge base. David Seiter, FNP-C, a recently appointed Associate Editor of JCAD, was invited by J.D.R. to lead the charge for this intermittent JCAD section, along with periodic contributions from others in dermatology. It is our hope that you find this short yet thorough article to be valuable enough that it is a “clip and save” article that you keep in your desk drawer or file cabinet in your clinic.
Cutaneous vasculitis is frequently encountered in dermatology practice and is often limited to the skin. However, a subset of patients may have associated systemic involvement affecting the renal, gastrointestinal, pulmonary, or neurologic systems. Distinguishing self-limited cutaneous disease from potentially serious systemic vasculitis can be clinically challenging, particularly in the absence of formal diagnostic guidelines. This brief report reviews factors associated with systemic involvement, including constitutional symptoms, gastrointestinal complaints, hematuria, and ulcerative or necrotic lesions. The role of laboratory evaluation, histopathology, and direct immunofluorescence in guiding risk stratification and targeted workup escalation is also discussed. A focused, evidence-based approach may help dermatology clinicians reduce diagnostic uncertainty while avoiding unnecessary testing. KEYWORDS: Cutaneous vasculitis, small-vessel vasculitis, leukocytoclastic vasculitis, systemic involvement, risk stratification
Introduction
The sudden appearance of cutaneous vasculitis presenting as palpable purpura, especially when it is extensive, often causes considerable concern to both patients and clinicians. Most cases are limited to the skin and resolve within weeks to a few months without sequelae, leaving dermatology clinicians to spend more time providing patients with comforting reassurance than working up the cause or treating it.1 However, cutaneous vasculitis may sometimes manifest as organ- and even life-threatening systemic vasculitis and/or may be associated with an underlying renal, gastrointestinal, pulmonary, or neurologic disease. Without clear guidelines for when and how to work up cutaneous vasculitis, many clinicians are left to worry silently along with their patients.
Dermatology clinicians most often encounter leukocytoclastic vasculitis (LCV), a form of small-vessel vasculitis (SVV) that is frequently triggered by transient or subclinical (often labeled idiopathic) exposures such as infections (eg, staphylococcal, streptococcal), vaccinations, or short-term medications (eg, antibiotics, nonsteroidal anti-inflammatory drugs). Persistent cases may be associated with chronic infections (eg, hepatitis B or C, HIV), long-term medications (eg, thiazides, anticonvulsants, allopurinol), autoimmune disease (eg, systemic lupus erythematosus, rheumatoid arthritis, Sjögren syndrome, inflammatory bowel disease) or innate immune dysregulation, and less commonly malignancy (<5%, most often hematologic).2 Other SVVs include immune complex–mediated entities (immunoglobulin A [IgA] vasculitis, cryoglobulinemic vasculitis, hypocomplementemic urticarial vasculitis), antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitides (granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis [formerly Churg-Strauss syndrome], microscopic polyangiitis), and antiglomerular basement membrane disease, all of which may have systemic involvement.
How to approach evaluation of SVV. Scanning the medication list for common triggers is a quick, low-cost intervention that can be done for every new SVV presentation, but knowing when to treat symptomatically with watchful waiting vs initiating workup for systemic involvement can be a murky clinical conundrum. Due to its heterogeneity, vasculitis has a classification system (the Chapel Hill Consensus Conference Framework) but not a formal, prospectively validated guideline for workup. While indiscriminate testing in low-risk disease may be discouraged, especially in the era of cost containment, clinicians may remain uncertain as to what counts as “low risk.”
To identify factors associated with systemic involvement, researchers associated with the Departments of Dermatology at Brigham and Women’s Hospital/Harvard Medical School and the Perelman School of Medicine at the University of Pennsylvania conducted a case-control study comparing 87 patients with systemic disease to 343 skin-limited controls.3 Their multivariate analysis published in 2025 found that the following factors were significantly associated with systemic involvement:
- Nausea/vomiting (odds ratio [OR]: 3.92),
- Ulcerating or necrotic skin lesions (OR: 3.26),
- Abdominal pain/cramping (OR: 2.71),
- Fatigue/malaise/lethargy (OR: 2.61), and
- Hematuria (OR: 2.40).
On univariate analysis, additional factors included:
- Joint pain (OR: 2.54) and
- Younger age (OR: 0.73 per SD increase [17.7 years]), likely reflecting the epidemiology of IgA and ANCA-associated vasculitides which are commonly systemic.
Patients with 1 or more of these factors warrant basic laboratory evaluation, starting with a complete blood count (CBC) with differential, complete metabolic panel (CMP), erythrocyte sedimentation rate (ESR)/C-reactive protein (CRP), and urinalysis (UA).4 Persistent, recurrent, or treatment-refractory vasculitis—even in the absence of the above features—also warrants prompt evaluation, as extracutaneous involvement may precede history and physical examination findings. For example, a UA and serum creatinine level can detect vasculitic renal involvement before hematuria becomes visible.5
While a core laboratory panel consisting of CBC, CMP, ESR/CRP, and UA should be obtained for all patients who exhibit features suggestive of systemic involvement as outlined above, dermatology clinicians may pursue further workup based on history, presentation, and biopsy findings.
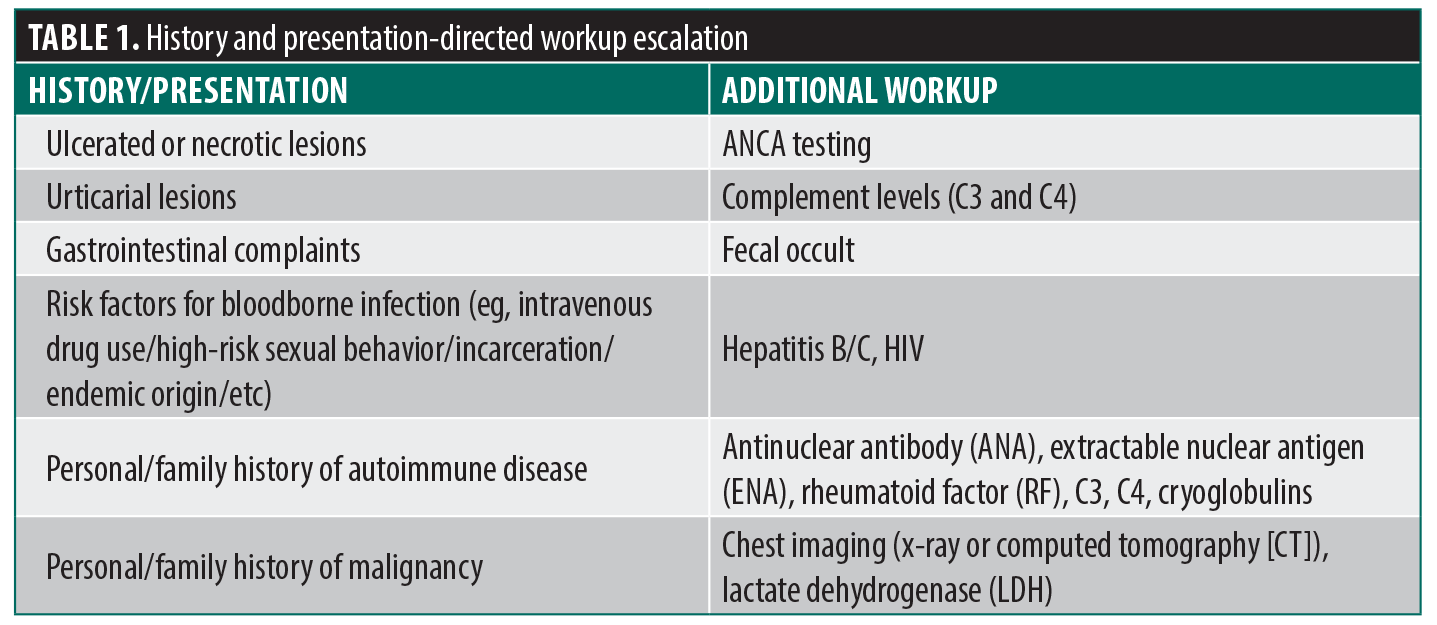
History and clinical presentation provide the first opportunity for targeted workup escalation (Table 1).
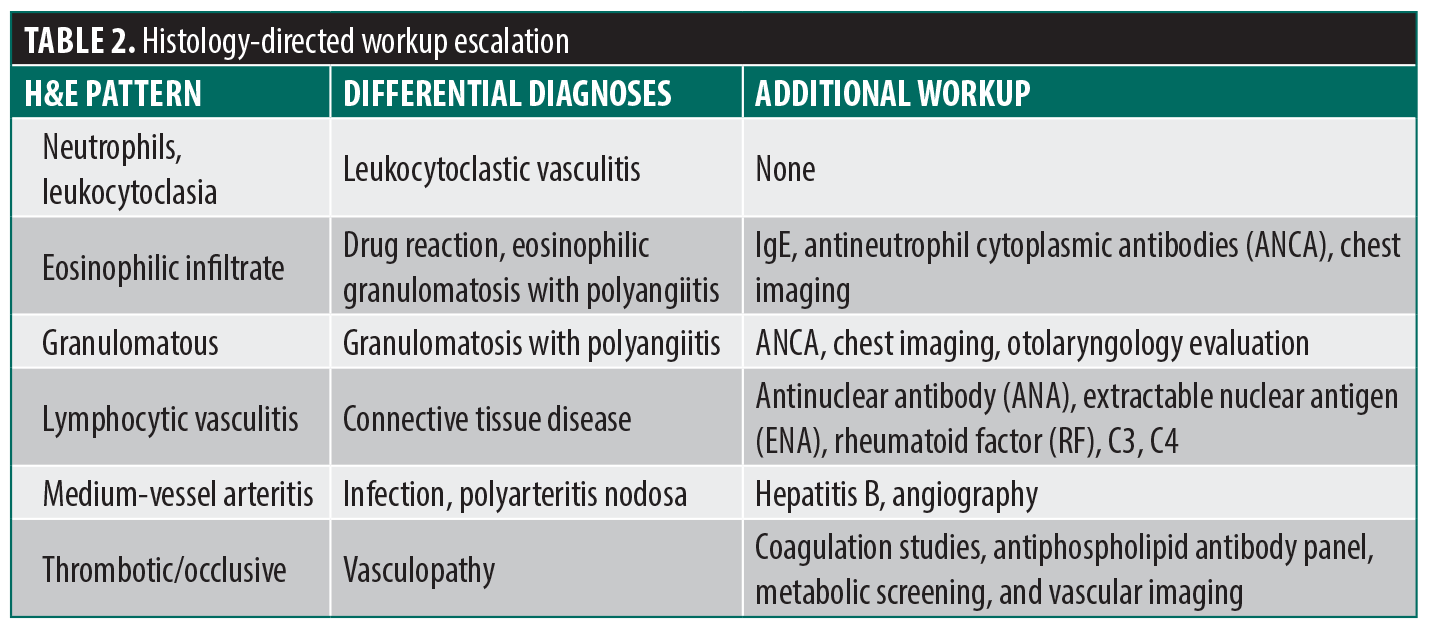
Skin biopsies for standard hematoxylin-eosin (H&E) staining and direct immunofluorescence (DIF) can also narrow the differential and direct downstream testing. Routine histopathology confirms the presence of true vasculitis by distinguishing it from pseudovasculitis (eg, occlusive vasculopathy, coagulopathy, or calciphylaxis), characterizes the inflammatory pattern, and, when the sample includes the deep dermis or subcutis, can identify medium-sized vessel involvement, guiding further workup (Table 2).
DIF provides additional clues to the underlying mechanism and guides targeted workup escalation in patients with clinical or laboratory features suggestive of systemic involvement (Table 3).
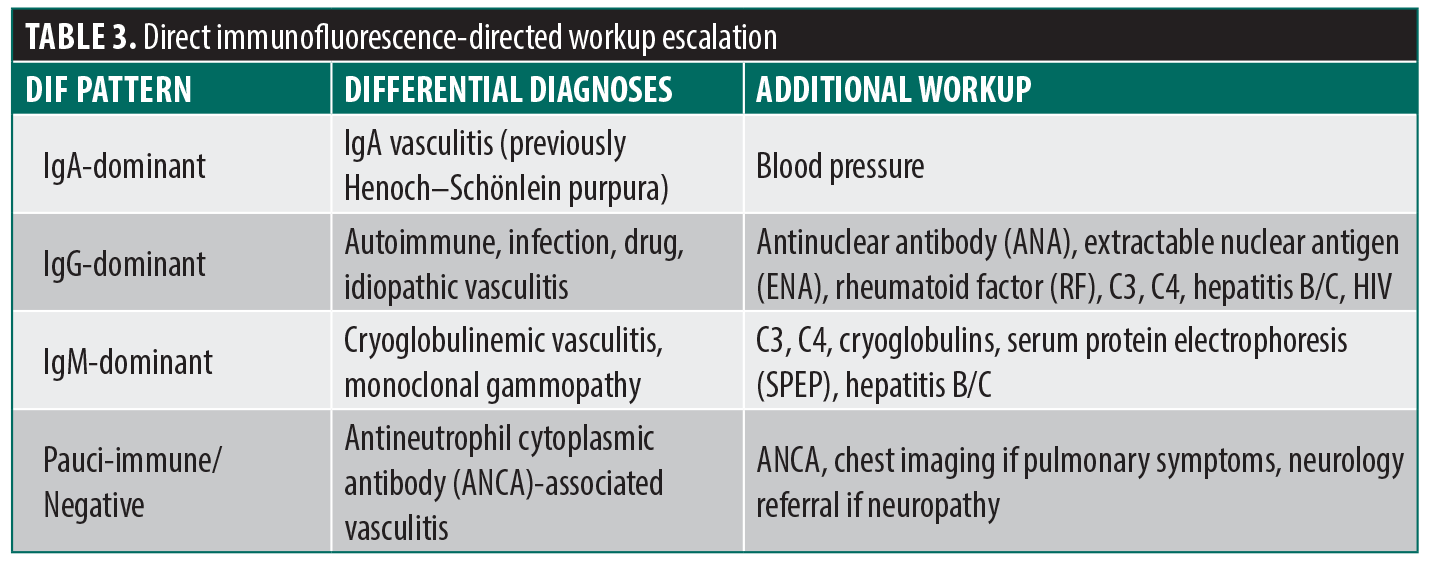
DIF biopsies provide the highest yield on new lesions (ideally 8–24 hours after appearance but no more than 48 hours) as immunoglobulins degrade rapidly, and they are best performed prior to topical or systemic corticosteroid exposure to avoid false negative results.6 It is important to punch the purpuric portion of the lesion for both H&E and DIF biopsies, not ulcerated/necrotic tissue or normal-appearing perilesional skin as required for DIF biopsies in bullous disorders because the immune complexes are confined inside the inflamed vessel wall.
When to refer? Dermatology clinicians may choose to refer patients for investigations not commonly performed in dermatology settings. However, a review of systems that assesses constitutional, gastrointestinal, urinary, and musculoskeletal symptoms and a focused physical examination to identify atypical or severe cutaneous findings are appropriate for every presentation of cutaneous SVV. If features with high ORs for systemic involvement are found, it is important to expedite basic serologic testing. Additional targeted workup as indicated by initial laboratory results as well as history, presentation, and biopsies may be subsequently ordered or referred to other specialties.
Concluding remarks. Findings from the multivariate and univariate analyses above provide clinicians with objective, point-of-care data to improve risk stratification and clinical decision-making for a common yet potentially serious dermatologic presentation.3 Evaluating cutaneous vasculitis for features associated with systemic involvement at the first visit, or at least early in the course of the condition, may reduce clinical uncertainty, diagnostic delay, and disease progression without the burden of overtesting. In the absence of these risk factors, observation with supportive care and close follow-up is reasonable.
References
- Baigrie D, Crane JS. Leukocytoclastic vasculitis. StatPearls [Internet]. StatPearls Publishing; 2023. Accessed 4 January 2026.
- Kermani TA, Warrington KJ, Amin S. Malignancy risk in vasculitis. Ther Adv Musculoskelet Dis. 2011;3(1):55–63.
- Mahajan A, Song W, Walls AC, Mostaghimi A, Micheletti R, Piette EW. Point-of-care risk factors for systemic disease in patients with small vessel vasculitis of the skin. JAMA Dermatol. 2026;162(1):94–97.
- Alpsoy E. Cutaneous vasculitis: an algorithmic approach to diagnosis. Front Med (Lausanne). 2022;9:1012554.
- Roper T, Salama AD. ANCA-associated vasculitis: practical issues in management. Indian J Nephrol. 2024;34(1):6–23.
- Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology. 2010;56(1):3–23.