J Clin Aesthet Dermatol. 2026;19(6 Suppl 2):S4–S14.
by David G. Cotter, MD, PhD; Brent Moody, MD; Jesus Vera Aguilera, MD; and John T. Vetto, MD
Dr. Cotter is with Las Vegas Dermatology in Las Vegas, Nevada, and University of Nevada Las Vegas School of Medicine, Las Vegas, Nevada. Dr. Moody is with the Skin Cancer and Surgery Center, Nashville, Tennessee. Dr. Vetto is with the Division of Surgical Oncology, Oregon Health & Science University School of Medicine, Portland, Oregon. Dr. Vera Aguilera is with Minnesota Oncology, Maple Grove, Minnesota and the US Oncology Network.
Cutaneous melanoma (CM) intersects dermatology, surgical oncology, and medical oncology. Recent advancements in melanoma diagnostics, prognostics, and therapeutics emphasize the importance of effective handoffs between these specialties in the care of CM patients. Tumor board discussions elevate care in both community and academic settings. Herein, we present a tumor board-like, expert panel discussion of 2 clinical cases of (CM). Keywords: Melanoma, gene expression profiling, sentinel lymph node biopsy ctDNA, 31-GEP
Introduction
Cutaneous melanoma (CM) is the fifth most common cancer in the United States (US). In 2026, there will be an estimated 112,000 CM cases and 8,510 deaths from CM. The 5-year survival of all comers with CM is 94.7%.1 When evaluated by stage, earlier stage at diagnosis correlates with improved melanoma-specific survival (MSS). However, an unacceptably high number of deaths occur in patients initially diagnosed as American Joint Committee on Cancer (AJCC) stage I. For example, T1a, T1b, and T2a melanoma patients have 98%, 96%, and 92% 10-year MSS, respectively.2 Approximately 70% of CM patients are diagnosed with T1a-T2a tumor,2,3 resulting in an anticipated 78,400 stage IA and IB CM cases in 2026. Hence, the paradox of early melanoma emerges: while the population performs well, any given individual may do poorly, and with nearly 80,000 stage IA-IB CM cases in the US annually, an unacceptably high number of deaths occur annually in patients that were originally diagnosed as early stage.
The Promise of Molecular Tools
Scientific advancement promises to transform the CM diagnostic, prognostic, and therapeutic landscape. For example, augmented intelligence (AI)-enhanced mole mapping, handheld clinic-based devices to detect melanoma, tape strip tests, and pathology based molecular tool all support earlier diagnosis of CM.4,5 From a prognostic standpoint, multiple tools are now clinically available, including 2 gene expression profile (GEP) tests and a circulating tumor DNA (ctDNA) test for CM. A prognostic 31-GEP can predict an individual patient’s recurrence free survival (RFS), distant metastasis free survival (DMFS) and MSS.6 In fact, the 31-GEP outperforms AJCC staging when it comes to risk stratification of AJCC stage I patients.7 The 31-GEP was also developed and validated to predict an individual patient’s likelihood of sentinel lymph node biopsy (SLNB) positivity.8 Furthermore, the 31-GEP test has been shown in numerous studies to be able to decrease the number of unnecessary SLNBs by about 20% to 30%.9–13 Recently, an additional GEP test has become clinically available. It is an 8-GEP that combines clinicopathologic factors into its risk assessment (CP-GEP).14 Both 31-GEP and CP-GEP have been studied prospectively. Both tests identify a group of AJCC low risk patients with an occultly elevated risk of SLNB positivity.14,15 In the DECIDE study, the 31-GEP identified a group of stage IB patients with an 18.5% SLNB positivity rate,15 and in the MERLIN_001 study, CP-GEP identified a group of stage IB patients with an 18.3% SLNB positivity rate.14 Despite the similar rates of identification of occult high-risk patients by both tests, only the 31-GEP identified a low-risk subset of stage IB patients (1.4% SLNB positivity rate by 31-GEP vs 6.5% SLNB positivity rate by CP-GEP).14,15 ctDNA offers an additional prognostic and surveillance tool. Patients with ctDNA post-resection have worse prognoses and re-emergence of ctDNA may indicate molecular recurrence before radiographic detection.16,17 Together, molecular and AI-tools have advanced our ability to diagnose, risk stratify, and surveil CM; nonetheless, meaningful changes in MSS remain elusive.
The Benefits of a Tumor Board
The melanoma therapeutic landscape continues to evolve rapidly. Notable changes in recent years include the approval of immune checkpoint inhibitors (ICI) for stage IIB and IIC CM based on the results of KEYNOTE-716.18 Additionally, data around the risks and benefits of neoadjuvant therapy, adjuvant therapy, and 1- or 2-drug ICI therapy continues to evolve.19 The rate of advancement makes it difficult to remain aware of all new updates as they emerge. National Comprehensive Cancer Network (NCCN) guidelines offer helpful guardrails,20 yet 8,510 people are still expected to die from CM in 2026.1 Certain clinical scenarios remain challenging:
- Identifying occult high risk stage IA to IIA patients
- When to perform a SLNB in T1b patients
- Management of SLNB negative patients
- Management of stage IIB and IIC patients
Issues surrounding these scenarios include appropriate use of prognostic GEP, SLNB, surveillance imaging, ctDNA, and adjuvant or neo-adjuvant ICI. Challenging cases may benefit from multidisciplinary tumor board discussions. Herein, an expert panel (BM, JV, and JVA) discusses 2 challenging clinical cases in a roundtable/tumor board style with special attention to recent advancements in melanoma prognostic testing and therapeutic intervention. Both patients provided informed consent and photo consent for this discussion.
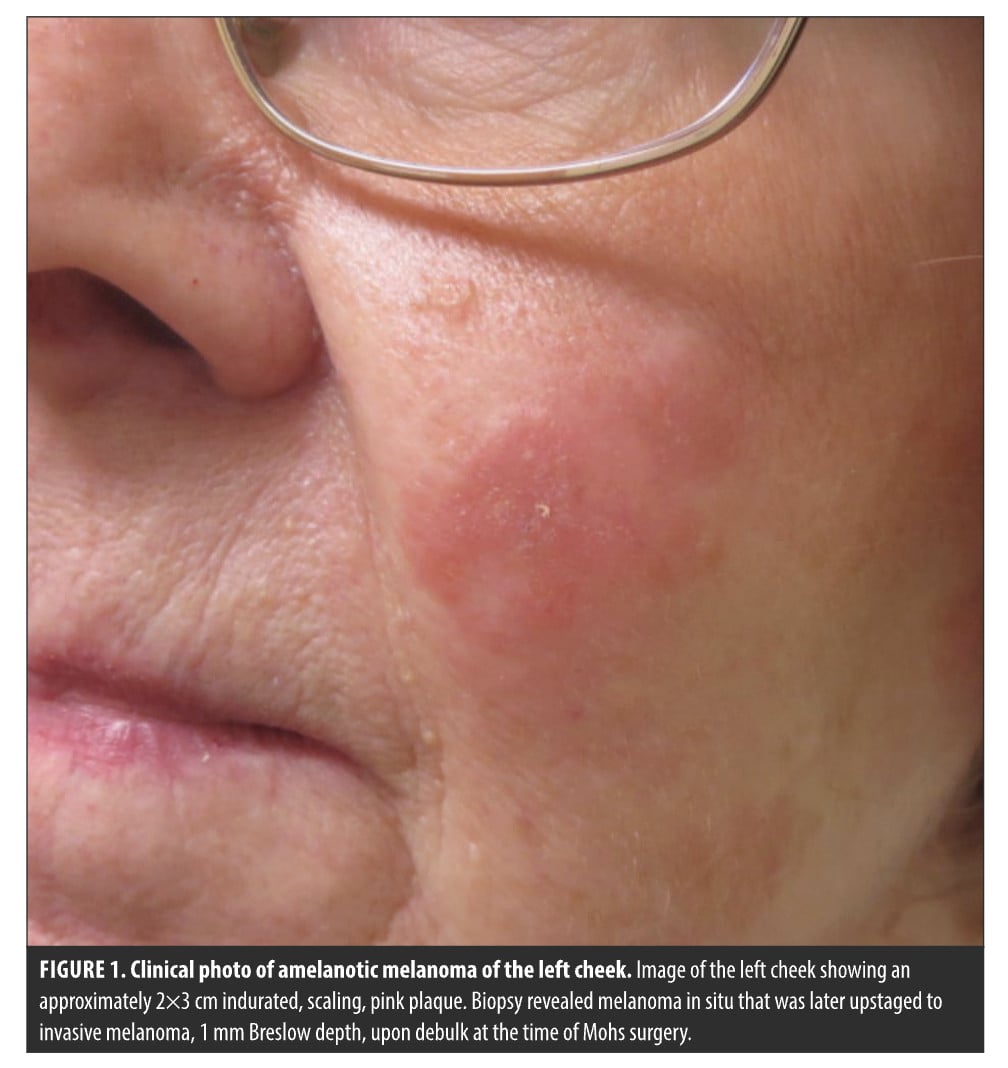
Case 1: To Node or Not to Node—74-Year-Old Woman With Amelanotic Melanoma
David Cotter, MD, PhD: We’re going to talk about a case that I like to refer to as “to node or not to node, that is the question,” particularly, for my patient here, who had a T1b melanoma. She presented as a new patient with a 4-year history of a 2×3 cm, indurated pink plaque on her face for 4 years (Figure 1). She told me it had not improved despite triamcinolone, clobetasol, tacrolimus, intralesional Kenalog, and even a prednisone taper. Her new primary care doctor thought this might be an autoimmune disease and had ordered an antinuclear antibody (ANA) test. Her ANA came back positive with an Scl-70 antibody, which is loosely associated with scleroderma. Certainly, the photo of this plaque is not at all consistent with scleroderma, but in my mind, I’m already starting to think about inflammatory skin diseases, perhaps tumid lupus, granuloma faciale, etc. When we’re thinking about inflammatory diseases, we like to do punch biopsies, not broad shave biopsies, and that’s exactly what I did. I did a punch biopsy into the center of this plaque to rule out tumid lupus. Two days later, my pathologist calls me to tell me it was melanoma in situ. In our practice, we have the ability to do Mohs with immunohistochemistry for the margins after debulking the tumor. Thus, a fellowship-trained, board-certified Mohs surgeon debulked the tumor, sent that off for permanent sections, and then did Mohs on the margins, cleared the melanoma, reconstructed it with a large rotation flap, and then 2 days later, we get the final pathology report that shows a 1-mm nonulcerated melanoma, 2 mitoses per high powered field, and no lymphovascular invasion. With that, let’s bring this case to tumor board. Now what?
Brent Moody, MD: I think, clearly, the primary tumor has been dealt with. We’ve had complete tumor extirpation and reconstruction. I think that’s settled. I don’t think you need any additional surgery at the primary site. Really, it’s what comes next, and the choices range from clinical observation, exams and nodal exams, all the way to consideration of SLNB. This patient at 1 mm would certainly qualify. I prefer the mitotic rate be 0 or 1, not 2. I think this case could go in a lot of different directions.
DC: Great, thanks for getting us kicked off there, Dr. Moody. Dr. Vetto, has this tumor been fully extirpated? She did not get 1-cm margins. She had a debulk with Mohs on the margins that showed clearance of the tumor. What’s next and does she need a SLNB?
John Vetto, MD: This patient didn’t get a standard wide excision. Of course, the current recommendation is 1 cm, and even the MelMarT-II (Melanoma Margins Trial II) will not go lower than that. We have a study here at OHSU where we’re looking at smaller margins, and that’s being run through our Mohs center. At this point, I would turn to my Mohs surgeons, and ask if they’re comfortable with that margin. If they’re not, we can re-excise it.
Regarding the sentinel node, there are 2 questions. First, can you find it? And secondly, do you want it? The first answer, for can you find it? Maybe not. There’s data from Gershenwald et al21 from MD Anderson that show that after an attempted Mohs, if you leave it open, after the central debulk, you can often still find the node, but after flap or graft, it’s harder, and you often can’t find a node. I would counsel this woman that we might not be able to find the node anymore. If she was strongly pushing for a SLNB, we could do a lymphoscintogram. Occasionally, a few times a year, I’ll get just a lymphoscintogram by itself—that is, not on the day of a planned sentinel node surgery, but just get it ahead of time and see if we can still see a sentinel node. If we either can’t find it, that is, if the dye doesn’t localize, or it goes all over the neck, then that’s no longer a technically findable sentinel node. So that’s question number one, can you find it? Probably not.
The other question, which I think is more important is should you even care? I ran a quick nomogram on this lady, using my favorite nongenetically based nomogram, which is the Melanoma Institute of Australia nomogram,22 which uses the clinicopathologic information you have already listed here. You plug in her age, absence of lymphovascular invasion (LVI), thickness, absence of ulceration, site, mitotic rate, and histology into the nomogram, and you end up with a 6% risk of positive sentinel node, which is well below the NCCN cutoff, which has just moved to 10%. Eight percent is my personal cut off, and in someone who’s highly motivated, but 6% is in the so-called “discuss and consider” range, and with a node that I might technically not be able to find anyway.
I’d therefore counsel her not to do the sentinel node, and if she’s worried about that, we can always do what we call “watchful waiting.” Watchful waiting is what we used to do before we invented sentinel node biopsy and involves examining her neck clinically and with an ultrasound every 4 to 6 months for 2 to 3 years. That’s what we used to do, and that’s what we do now post-MSLT-II. If she’s really worried about nodal involvement, but I don’t feel the point in going after it, and my Derm surgeons tell me that she’s adequately excised, we can do some watchful waiting, with a 94% chance that we won’t find anything.
DC: Excellent. Thanks for that perspective. Dr. Vera, what are your thoughts if you are seeing this patient?
Jesus Vera Aguilera, MD: I don’t see many of these, but I do get some of these cases. If you pull the data from the MERLIN trial, you’ll use it to guide your decision as to whether you should do a node biopsy or not. Until recently, the NCCN guidelines did not recommend incorporating GEP into clinical decision making. For the question of SLNB, it’s a valuable test, and in some cases, I order GEP, and then I have to decide which additional resources to use: computed tomography (CT) scans, ultrasounds, positron emission tomography (PET) scans, ctDNA, and/or DecisionDx. For this specific case, assuming the node was unable to be found and/or that it’s negative, I would consider using ultrasound. It’s a very sensitive tool to find earlier occurrences. We’ll talk more about the ctDNA. There has been a new boom about what to do in these early melanomas. What’s the use of circulating DNA? It’s a great tool which also has good data for other tumors, such as colon cancer, for example, but does not have a lot of data in melanoma, and most of the data is for stage III or stage IV. Once they have been resected, if you have a positive ctDNA, there is a higher likelihood of recurrence. But when you have a stage I or stage II, it’s a really difficult area to navigate. This is a great area for discussion of what we do in the community.
DC: That was a nice overview of some of the molecular tools that we can utilize to help prognosticate and also monitor our patients. Let’s unpack the question around GEP and using it as a tool to either rule in or rule out doing a SLNB. Let’s assume that she hadn’t already been reconstructed, and we knew this was an invasive melanoma from the get-go. How are we approaching any given patient in the T1b range, and what is the utility of GEP? Dr. Vera mentioned 2 different tests, DecisionDx-Melanoma and the Merlin test. I think there’s a lot that we can discuss there.
JV: So let me start. First of all, let’s say that she hadn’t been resected by Mohs, didn’t have a flap or graft, and we knew up front that she had 1 mm with 2 mitoses, so we would consider her for SLNB. To estimate the risk of a positive sentinel node, I’ve already mentioned non-genetic-based nomograms, but surgical oncologists often start with the good old “Rule of Thumb” where you just move the decimal point over from the Breslow and add 5% for ulceration—it’s not a very good rule—but if you use that, her risk becomes 10% and if her performance status is good and she’s motivated, I might do the standard wide excision and SLNB on her.
If I thought that it would change management by incorporating molecular testing, then we could order the DecisionDx, 31-GEP. That test gives us the individualized risk of SLNB positivity, which is at least as good as the nomograms. If that came back high, say more than 10%, that would push me to do the sentinel node. In terms of the Merlin CP-GEP test, the data are odd: the NCCN just quoted the Merlin_001 study, but they did it in a funny way. They noted that the study was originally designed to show that a low risk profile would predict a <5% likelihood of SLNB positivity, but, in the study, Merlin low risk patients unfortunately had a 7% likelihood of SLNB positivity. Then, the NCCN said, if it’s less than 10%, you can use it, so they quoted the data, but they didn’t quote it the way it was written. Then in the following paragraph, they say these tests should be looked at with prospective studies, which is exactly what we’re doing here at OHSU. When we run a Merlin, we run it on a prospective study. We have a prospective study here at our university where patients who are Merlin low risk by the new NCCN criteria, meaning less than 10% don’t have a sentinel node, and we follow them with ultrasound. We also study the 31-GEP prospectively and have published that data.
That test is a little broader based. It has the SLNB prediction tool in it, but it also has the risk of recurrence (ROR) tool, and I like to use that for decisions made past the sentinel node. For example, if the sentinel node is negative and the stage is low, what do you do next? In those cases, I use the 31-GEP ROR to determine if the patient is high risk or low risk for recurrence and guide my follow up accordingly. Our own data, which is in press, shows that if the 31-GEP is low risk, Class 1, and the stage is AJCC low risk, stages IA-IIA, we have no recurrences in 100 of those patients so far. I like the test for that purpose. The flip side of it is if the patient’s sentinel node negative, stage Ib in the case presented, but the patient has a high-risk 31-GEP, in our data, that’s where all the recurrences, distant recurrences, and all the deaths occur.23 I’d like to see those patients (node negative, 31-GEP high risk) go forward to either get ctDNA or adjuvant therapy on a study. That has to be done prospectively, just like the NCCN says.
DC: Excellent. I think that’s great perspective and a high-level overview of the utility of both tests and some of the difficulties of the way the Merlin_001 study was quoted in the NCCN guidelines. Dr. Moody or Dr. Vera, do you have any other thoughts about the utility of GEP in these types of cases?
BM: I’d like to touch on one other thing that was mentioned earlier, and that would be the role of ultrasound. NCCN guidelines currently don’t really push for a lot of imaging in these patients. But our European colleagues, in their guidelines, really recommend ultrasound for all stage IB, and higher. I think ultrasound certainly would be very appropriate in this patient. This is the type of patient that our group really does lean on the gene expression profiling. As you mentioned, it can definitely rule-in a lot of things. If someone has an unfavorable profile that is going to push us to consider SLNB. It’s certainly going to consider more aggressive upfront imaging beyond ultrasound, and also consider ctDNA. The 2 data points that we get from i31-GEP that would be relevant here are the sentinel lymph node prediction model and RFS. We have decided as a group that patients with a RFS of 90% or less, are considered in the high-risk group. If she fell there, she would likely be followed with ctDNA, as well as more intense surveillance.
DC: Let’s talk about intense surveillance, and then the utility of imaging. What do you guys consider appropriate serial imaging for a high-risk, node-negative patient?
JVA: If you quote the NCCN guidelines, for this specific case, we should perform history and physical (H&P) every 6 to 12 months for 5 years, blood tests not recommended, imaging not recommended, just as needed or if the patient becomes symptomatic. However, in my practice, for high risk GEP patients, I usually recommend baseline cross-sectional imaging followed by ultrasound in the area SLNB was performed 3 months later, then I repeat cross-sectional imaging at 6 moths, and ultrasound at 9 months. I complete this work up with another cross-sectional imaging with either CT scan or PET/CT 12 months after surgery. I tend to do this in the first 2 years after resection, because that’s where I see most of my patients having recurrences.
DC: I just want to make sure I understand: it’ll be a 3-month node ultrasound after the surgery. At Month 6 post-op, it would be a CT scan or PET scan, another ultrasound at Month 9, and then at the 12 month mark, another high resolution scan. That gets you through the first year and maybe you repeat that out to Year 2, depending on the patient, and you might be layering in ctDNA along the way for your highest risk patients.
JVA: Yeah, exactly. That’s how I do it in my practice. I would be interested to see how the rest of the crew does it.
JV: Let me address ultrasound, and then cross-sectional imaging. We use ultrasound, in 2 different settings. First, if a SLNB-eligible patient refuses a sentinel node or can’t get a sentinel node for medical reasons, or if you inject the radio tracer and there’s no uptake, we followed them with watchful waiting, as I’ve already described. That is, instead of going to the OR, we have our Derm surgeons perform the wide excision under local and then we follow the nodal bed with ultrasound. It’s not perfect, but for the reasons I’ve mentioned, that approach is sometimes all we can do in SLNB-eligible patients, and it is actually used in some countries instead of SLNB. I do not think it’s as good as SLNB, but if it’s all you got, that’s what you do. The second reason we use ultrasound is after a positive sentinel node. There are patients who will sometimes fail only in the nodal bed and we will do ultrasound independent of cross-sectional images to catch that as early as we can. That is a recommendation that came out of the Multicenter Sentinel Lymph Node-2
(MSLT-2) trial.
To get back to the case, she’s AJCC, low risk, but let’s say that her 31-GEP comes back high risk. She’s class 2A or 2B, now what? Unfortunately, we don’t have any prospective data for those patients to treat them with adjuvant therapy, but what you’d like to do, at the least, is follow them a little more closely. To examine this, we ran a study between 3 centers, OHSU, Cleveland Clinic, and Northwestern where we looked at exactly those patients: patients who are AJCC low risk (IA–IIA) but GEP Class 2, and we scanned them every 6 months per NCCN guidelines for higher risk melanoma.23 We showed that we detected recurrences a year earlier compared to patients that were not being scanned. Scanned patients got immunotherapy more often because they had better performance status, and the result was that scanned patients had a better survival. Now, that was nonrandomized, 3-institution, prospective selected data. I’m trying to convert that into a prospective study. It’s proving difficult, but I think that our study is at least proof of principle that patients who are high risk genetically should be followed by NCCN guidelines for high-risk patients, and that is with scanning. What we’re doing at Oregon is basically redefining what it means to be high risk and expanding the definition to include either high risk by AJCC staging and/or high-risk biogenetic expression.
BM: What you’re trying to do is identify those patients at high risk for disease progression. What is a patient’s pathway to being put in the high-risk category? It can be traditional staging, or it can be biomarkers, like GEP. We image at baseline all our high risk GEP patients. For our highest risk GEP patients, it’s the first thing we do. We PET-CT all those patients and have a multicenter prospective trial going on, currently looking at the utility of that. In quite a few of the patients we image, we find actionable items that turn patients from surgery plus adjuvant immunotherapy to neoadjuvant therapy. We lean very heavily on the GEP to guide some of those imaging decisions.
DC: Great. Thanks for that perspective. We did order the i31-GEP. It came back as a class 1A. The GEP score was 0.25. Her likelihood of having a positive sentinel node was 4%. Her RFS was 90.1%, which is right above Dr. Moody’s threshold of 90%. Her DMFS was 93.7% and her MSS was 98.4%. With that in mind, are we happy with routine follow up in general dermatology, or are we doing more?
BM: I’ll answer for dermatology, since you started here. I think in this case, that’s a favorable genetic profile… I think we would probably just put her in the lower risk category and she would have clinical follow up.
JVA: I do see many referrals like this to my clinic. When you went over the [GEP] results, I felt better about the case. The clinical features originally made me uneasy (location, presentation), but the patient has a low risk for recurrence. Based on recent data regarding favorable risk GEP, one could consider following with dermatology alone. While GEP shouldn’t be used to make clinical decisions, I think it is a great tool to complement standard of care.
JV: Those numbers are very encouraging. This is the type of patient that I would call “double negative.” That is, they have low risk AJCC stage and their GEP Class is 1A. In our data here at Oregon, we don’t have any distant recurrences in those patients, only 2 loco-regional recurrences. To me, loco-regional rucurrences are technical, not biologic, problems, and they can be easily picked up by dermatology. She can be followed by derm. If the providers want to get a baseline ultrasound, I think that’s fine.
DC: I’m glad to hear that because that’s exactly how we managed her. She had low risk derm follow up with routine skin checks, and she’s been disease-free for almost 5 years now.
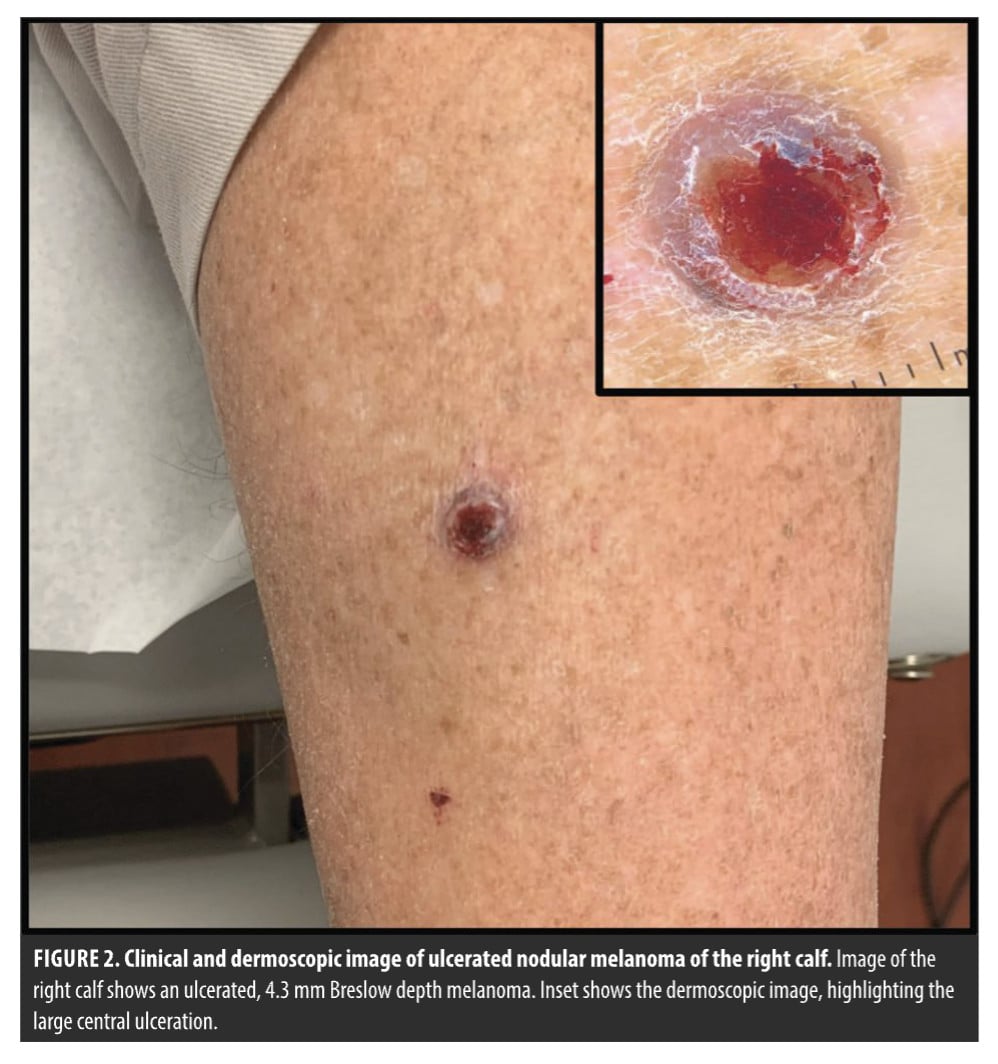
DC: This is the case of an 83-year-old, asplenic man who came to me as a new patient with this ulcerated nodule on his right calf (Figure 2). A diagnostic excision with 1 mm margins came back as a 4.3-mm ulcerated melanoma, with 3 mitoses per high powered field. What do we do with this T4b melanoma? Wide local excision, SLNB, neoadjuvant therapy, adjuvant therapy, imaging, and/or ctDNA? The answer today may be different than what the answer was for this case a few years ago. Nonetheless, why don’t we start with the immediate next steps for this patient. If you were seeing him today in your practice, with all of the new tools, tech, and data we have available, what would you do?
JVA: I would be really worried about metastatic disease from the get-go in this case. Whenever I see a pT4b ulcerated melanoma on the legs, the first thing I’ll do, before even talking to my plastic surgeon, will be to do cross-sectional imaging. Then the next question, is the node positive or negative? If the node is positive and no metastatic disease is seen, based on the NADINA trial, we would prefer to proceed with neoadjuvant immunotherapy treatment.24 Of course, we have to consider the patient performance status and the patient’s wishes. I’m going to be giving quite a good dose of immunotherapy, ipilimumab and nivolumab, and some patients do very well, while some others, not so great. After 2 cycles, rescan, take the patient to surgery and remove the node, and see what was the response. Do we have a pathological complete response? Then we usually continue with nivolumab to complete the year. During adjuvant treatment and after this is completed, we ultrasound every 3 months in between cross-sectional imaging. The challenging cases are those patients with high-risk node-negative melanoma. Assuming this patient was node negative, he would be stage IIC, NCCN guidelines recommend adjuvant immunotherapy or radiation. The question is, do I need to do the adjuvant treatment? Am I going to cause harm? I’ll give you a couple of examples. If I have a high-risk patient that is node negative and the patient is motivated to receive treatment, I’ll proceed with immunotherapy, as we have very strong data for it. If the patient is not motivated for systemic therapy or if the patient has autoimmune disease or history of transplant, then I’ll consider frequent surveillance.
I’ll give you an example of a patient that came to my office last week with a stage IIB melanoma, but the [GEP] class was 1A, and the patient had ulcerative colitis. I thought, “I’m not going to put you through adjuvant treatment, but let me do frequent surveillance.” So far, she has felt confident based on her low GEP testing. I hope that things are going to go well. I would like see more data for stage IIB/IIC melanoma patients with low risk GEP, maybe we could avoid overtreatment and reduce imaging in some patients.
JV: I agree with everything Dr. Vera said. I would add to it. The newer terminology for this patient is “high-risk, node negative,” assuming they’re clinically node negative. That is, the AJCC defines stages IIB, IIC as high-risk node negative. You have to scan these patients right off the bat. All the guidelines say that—the NCCN says that, the European guidelines say that. The other new term for these patients is they are “drug eligible,” meaning that the US Food and Drug Administration (FDA) has approved immunotherapy for IIB/IIC based on the Keynote-716 CheckMate-76 trials. So here you have a patient who is at least drug eligible. But the question is, are they neoadjuvant eligible because the cats out of the bag and they already have nodal or distant disease? To know that, you scan them right out of the box, and you figure out if they already have a macro, nodal disease or distant disease, then you know that they need to get at neoadjuvant therapy.
Now, assuming the scans are negative, which they most often are, then the patient remains clinically high-risk node negative, and then they’re indicated for wide excision and SLNB. In such cases, I’m very anxious to get the wide excision and sentinel node done, because the sentinel node will tell us if he is actually stage IIIB/IIIC, in which case, he definitely needs adjuvant therapy. The ASCO and the SSO guidelines for doing sentinel node on a patient like this, who’s T4b clinically node negative, state that SLNB in such patients is “a consideration,” and that’s a little surprising to me. We’ve written papers through the Sentinel Lymph Node Working group that show that the sentinel node is still very valuable in in these patients,25 but I know that there’s been a lot of rumblings about skipping SLNB in these thick, ulcerated patients, and going straight to adjuvant therapy after the wide excision. That makes me nervous, because I’d like to know their actual pathologic stage, because that tells us how much we should push for the adjuvant therapy, assuming the scans negative. I believe that such an approach—going straight to adjuvant therapy without SLNB—creates a subgroup of patients that I see as a surgical oncologist where the sentinel node, for whatever reason, was not done, the patient goes on adjuvant therapy, and then they progress only in the nodal bed. Such patients are then referred back to surgical oncology, and we are asked to now do a larger dissection than we would have done if you done if they had had a positive sentinel node. In our center, we follow the ASCO/SSO “consideration” for SLNB, so that we stage patients better, and perhaps avoid isolated nodal progression later. Dr. Vera, what do you think? Is that what you do?
JVA: Yeah, I find the use of sentinel node biopsy very useful…but some school of thought will say, “Well, you have really good immunotherapy. Wait. It’s okay to just let the tumor grow.” But then I’m like, I don’t know if I can extrapolate that from other cancers. I will say, I want some of the disease, but I don’t want a big bulk of disease because that becomes challenging to manage. I think the SLNB gives me a lot of information. I know what to look for, I know where to ultrasound. I know exactly what is happening in the microenvironment, as well. I wouldn’t shy away just yet, let’s consider and discuss.
JV: And I’ll say it again: if you do the sentinel node up front, you’ve subjected the patient to fairly minimally invasive surgery. I mean, no surgery is noninvasive, and you do lose nodes, but if you skip SLNB and believe in the immunotherapy so much, and the patient progresses only in the nodes, now you’ve got macrometastatic disease and are subjecting the patient to a more formal node dissection, with a higher risk of lymphedema and long-term morbidity. I think that is another reason to follow the SSO/ASCO guidelines and stage these patients up front.
BM: I agree with what my colleagues are saying. I think there are several things to think about here. You know, we tend to, if this patient had demonstrable macroscopic disease on imaging, of course, we will want to do neoadjuvant. Typically we do the NADINA protocol. If there’s some hesitation with both drugs, SWOG S1801 showed that pembrolizumab works well as neoadjuvant as well. Neoadjuvant is certainly an option. In patients with macroscopic or radiographic nodal diseases, I think most of us would do that. Now, SLNB for this patient, lots of advantages, as have been mentioned. You get prognostic information, a decreased risk of nodal relapse. The patient would be eligible for targeted therapy upfront, whereas, if you didn’t have the SLNB, they would not be eligible for targeted therapy. I think those are all reasons to consider SLNB. I will tell you, we’ve had some patients who are committed to drug, regardless. This patient would be drug eligible. My personal view is that in the absence of some strong reason not to, all 2C’s probably should have adjuvant drug. For 2B’s, I think you can argue back and forth, but for 2C’s, that would be my preference… I could see where there might be instances where people would not do a SLNB if you’re going to go to drug anyway. I’m not sure what happened with this patient, but we’ve had a few patients that absolutely were drug eligible, and like, “I want drug. I don’t want to have the surgery. I just want drug.” And we’ve done that a few times. I wouldn’t say it’s a standard practice, but it’s something that we’ve done.
DC: It was obvious from the get-go that this was an aggressive, fast-growing melanoma. I was worried and ordered the i31 GEP, which came back as a class 2B. MSS was 80.5%, DMFS is 60.7%, his RFS is 36.9%, and his likelihood of SLNB positivity is 24.6%. With that information, the patient knew he would want to go on ICI therapy irrespective of the results of a SLNB. I conferred with a medical oncologist and surgical oncologist in town, and after discussion, all parties agreed to move forward with an in office wide local excision without a SLNB. He had baseline cross-sectional imaging, which was negative, and he went immediately on adjuvant ICI.
JV: You can argue it either way. The risk [of lymphedema] in the MSLT-1 trial was 1% for the SLNB, and the 1% tend to be patients with risk factors, vs the 25% chance this patient has a positive node. So that means that, without drug, there’s a 25% chance he will progress in the nodes. We don’t have the exact numbers of how much drug reduces the chance for nodal-only progression, but surgical oncologists still definitely see patients who are on active immunotherapy, but they’re sent back by medical oncology to ask, “Oh, they’re only progressing in the nodes, will you now do a node dissection?” and we say “Oh, we wish we’d done the sentinel node.” So you’re trading a 1% risk of lymphedema for risk of definite lymphedema, because now they need the node dissection. We know from the MSLT-2 trial, which was done before the era of immunotherapy, that in 25% of patients after SLNB only, tumor was left behind in the nodes, and that tumor persists and progresses. While drug reduces that, data from the Sentinel Lymph Node Working Group, the best data we could get, suggests it only reduces it to about 10%,26 so I think you’re trading a 1% risk of lymphedema for a 10% risk of lymphedema. So, I would have erred on the side of doing a sentinel node, but that’s because of my take on the numbers.
DC: And then just a question for the dermatology readership, in these types of scenarios where you choose to do a SLNB, does that provide a certainty that there won’t be nodal progression in these types of cases, or certainly you could still have nodal progression even after either a positive or negative sentinel node biopsy, and what are those rates, and how does that compare to not doing it and going straight to drug?
JV: That’s a great question. Anytime you do a sentinel node in anybody, whatever their AJCC stage turns out to be, there’s always a risk of what’s called a “false negative sentinel node.” We used to call it “missed sentinel node,” but that’s kind of a risky term to use around patients, so we call it a false negative, meaning you took the node out and it was negative, and everyone is happy about that, but oh, by the way, it wasn’t the actual the sentinel node. For whatever, often technical, reasons, the node that was removed was actually not the sentinel node, and that leads to a false negative result. A false negative result means that the patient who is SLN positive relapses in the nodal bed, usually about 19 to 24 months later, right underneath the lymph node scar, which means at the sentinel node procedure, you just left the correct node behind. That happens on average about 5% of the time. We like to say that sentinel node biopsy is 95% accurate, which sounds great, but you flip it around, it means there’s a 5% false negative rate. That false negative rate varies by the nodal bed; measured at our institution, it’s only about 2% in the axilla, about 5%-10% in the groin, but it can be as 15% or higher in the neck. The false negative SLNB rate is why some surgical oncologists get nervous about doing sentinel node in the neck, because of the higher false negative rate which is coupled with a lower positivity rate, by the way. So you’re absolutely right, Dr. Cotter, you have to balance all these numbers against the false negative rate, but the average of 5% is still lower than the risk of progression in the nodes.
DC: Thanks for saying that. I think that’s helpful for everyone in the JCAD readership to understand that those rates change depending on what body location. Even in a case like this, where we’re on a lower extremity, and we’re talking about potentially a nodal recurrence in the groin, if the false negative rate is a 5%, and doing the ICI brings us down to a 10% risk of nodal progression, we still have a 5% swing there. Thus, there may be some advantage to doing the sentinel node from the get-go then.
JV: You’re absolutely correct. By the way, how did the NCCN come up with the recommendation to not do the sentinel node if the risk of positivity is less than 5%? It is based on the 5% false negative rate, because if your risk of finding a positive node is the same or less than missing it, then the sentinel node procedure becomes a wash.
BM: Glad you mentioned that Dr. Vetto. It’s not something that, at least in the dermatology community, I think there’s a great appreciation of that the lower someone’s risk of a positive lymph node biopsy is, the more important that 5% false negative becomes. If someone’s positivity rate is, for example, 7% with a 5% false negative, that’s really close to equilibrium. On the other hand, this patient, his likelihood of positivity was 25%, so again, that spread becomes much less meaningful clinically.
JVA: And you have to add the percentage varies depending on patient motivation and performance status. Also going back to what Dr. Vetto mentioned, when you have a recurrence, you can also consider radiation to the area as well to achieve some local control.
DC: Excellent thoughts. He did go straight to ICI after a wide local excision with 2-cm margins. He did not have the node. He actually didn’t want to go to the OR and undergo general anesthesia. He had concerns about that. We ended up re-excising him within
2 weeks of his initial diagnosis. He went on ICI within 2 weeks of his initial diagnosis, single drug therapy. About 8 weeks into therapy, he developed autoimmune thyroiditis that was managed with levothyroxine, and then maybe about 6 months later, he progressed. He had some metastases in transit to the thigh that responded to ICI therapy. He was just left with melanophages when we rebiopsied him. Then he developed some bulky lymphadenopathy in the groin that was surgically managed about a year later. He did really well after that. Now he’s about 3 and a half to 4 years out and still doing absolutely fine. He’s been managed on ICI therapy throughout his entire course. But to your point, Dr. Vetto, I think you hit the nail on the head here in terms of balancing risk. Do you go straight to these checkpoint inhibitors? Is there additional advantage to doing the sentinel node to try to avoid local nodal progression? So now that you guys know the outcomes, I’d love to hear additional thoughts and what other tools you think might be in play for these types of patients.
JVA: I’ll say medicine, in retrospect, is very easy… [If] at the time [of diagnosis], the patient would have had a positive node, then I could have offered neoadjuvant ipilimumab/nivolumab. That way I would probably be more aggressive from the get-go to avoid that recurrence. Otherwise, for stage IIB and IIC, single agent adjuvant pembrolizumab and nivolumab are the ones that are approved. Dual immunotherapy with cytotoxic T-lymphocyte associated protein 4 (CTLA-4) and the programmed death-ligand (PD-L) combination is more toxic, but you see
better responses.
JV: Well, I’d like to ask my colleagues: the GEP was obtained here and it came back high risk (Class 2B), which is one of the criticisms in high risk, node positive patients. The arguments is often made that the GEP doesn’t add anything, but here it certainly pushes you, Dr. Vera, to treat. What about if it had come back Class 1? As you may know, SEER independently provided some data a couple years ago on approximately 12,000 patients who had been been tested with the 31-GEP, and of those, they’re able to extract 775 who currently would have been drug eligible, age were pathologically node negative, high risk (Stages IIB and IIC). All this data was extracted before the FDA indication came through for adjuvant pembrolizumab and adjuvant nivolumab in these patients. Here were 775 patients who hadn’t seen drug, and SEER was able to show that if they were (GEP) Class 1, they did very well. If they were class 2, they did not.27 Do you ever use the GEP, as we did in this case, to push for drug? Conversely, have you ever used it to de-escalate?
JVA: When you’re there with the patient and you have to make a decision, a life impactful decision, you want to get all the information you can. And one of these pieces of information is the DecisionDx. I have been using the DecisionDx test for the last 2 years, and I have node negative anecdotes from my clinic. Two of them were class 1A. I decided to just wait a little bit [knowing we could be aggressive if needed if we were to detect a recurrence on imaging]. Luckily for those 2 Class 1A cases, there have been no recurrences after 2 years. Whenever I see a class 1A, even if it’s a stage I, I feel really relieved. On the other side, I’ll flip the question back to you, what would you do for a stage 1a melanoma, stage 1A/1B, with a high [GEP] class? What’s your what’s your comfort in those cases?
JV: Yes, in those cases our medical oncologists don’t escalate therapy for clinical stage 1, low risk, and that makes sense. There’s no data for that. But as I mentioned, we do escalate imaging, because we all know that all these drugs, whether it’s immunotherapy, targeted therapy, whatever, all work better with lower volume disease. If you can catch disease asymptomatically on a scan, before the patient has a seizure or bowel obstruction or hemoptysis or some other terrible symptom, they’ll do better. All the data since the late 80s shows that, and with all the immunotherapy we’ve developed since that time, patients, as you know, do better with lower tumor volume. We want to scan those patients and catch them early, but we don’t have any data to escalate therapy in non-drug eligible but GEP high-risk patients. I think that would be a spectacular trial. For now, we’re just scanning them. I think that’s a safe move, and we’re doing that based on our retrospective data.
BM: Dr. Cotter, back to your original question, if this patient, or a similar patient, was low risk, I think it would really depend upon what is their predicted RFS? Because we give adjuvant drug in these instances to decrease recurrence. So if they have a very favorable GEP and a favorable RFS, how much upside potential do we have if their RFS is, you know, 85% and let’s say we can give them drug and push that to 92% or 90%, is that amount of benefit worth the risk and cost of the drug? I think that really comes into play in Clinpath stage IIB’s. A lot of those patients do really well with observation. I think it’s Clin path stage IIB with favorable genetics, a lot of those patients can be observed and just treated at progression. On the other hand, a IIC like this, generally we’re treating the IIC’s. On the other hand, if patient had a lot of hesitancy or had a relative contraindication to immunotherapy like Crohn’s that’s not terrible, but manageable, or things like that, things that just give hesitancy and they have a good genetic profile, it may make sense to observe that patient closely. I think even in these advanced melanomas, having that risk stratification is helpful. Having the additional piece of information never hurts. There’s no downside to having this additional piece of information, even in these advanced melanomas, and talking about what to do with some of the earlier patients, the patients that give me the most indigestion, are my stage IIA patients who are not drug eligible, but have a bad GEP, because they are ticking time bombs. We can’t give them drug to diffuse that time bomb. We just have to wait and hope it doesn’t go off. Those are the patients that we image, that we follow with ctDNA, that we really surveil closely. To Dr. Vetto and Dr. Vera’s point, if they do progress, we’re treating them as soon as possible. That’s why in those patients, having that GEP knowledge is helpful. It tells me who gets ctDNA and who I need to image? I don’t want to image and ctDNA a bunch of low-risk people. That’s doesn’t help them, and it’s a waste. Again, it really is a primary risk stratification tool, along with our traditional staging.
JV: I think it’s about resource management. We can use these genetic tests to figure out who we’re probably following too much and can just go back to derm only, and who we should be following more closely. I estimate about 10% of patients are discordant; they’re low risk stage, but high risk GEP, and resources should be shifted to those patients, like Dr. Moody says.
BM: A recent abstract from Ohio State University showed that 12% of pT1a melanomas do not have a class 1A GEP. That is, 12% don’t have the preferred result from i31 GEP and that’s significant. One in 10 of thin melanomas that we think should do well actually have not the best GEP profile. It happens.
JVA: It also happens that I get some stage I melanomas, class 1A and I’m like, well, thanks for the referral. I think your patient’s going to do just great. I don’t really need to see those, but if I see a high risk GEP, even it’s an early stage, I’m happy to help with surveillance, or to push for adjuvant treatment for the right patient. All of these things (ICI therapy molecular testing, SLNB), add cost to the care, which is not cheap. The GEP testing is not cheap either, but I think the SLNB is not as expensive. That’s what I heard.
DC: Great follow up on that Dr. Vera, I couldn’t tell if you were joking around, or you’re being serious with the cost of the sentinel node vs molecular testing.
JVA: I’m serious, like how expensive is it to get SLNB? I think it doesn’t add too much cost to the patients.
JV: Sentinel node biopsy is not free. I’ve seen estimates as high as $20,000 when you add everything, the trouble is you have to add all the bundled expenses, the surgical fees, path fee, the anesthesia fee, the recovery fee. All that adds up. The charge vs actual cost of these genetic (tests) can be hard to cull out… based on what actually is reimbursed, not charged… But Dr. Vera is right, if you have a negative sentinel node in a IIB/IIC patient, so they don’t upstage, and they decide that they don’t want adjuvant therapy based on the Checkmate 76 or Keynote-716 trials, then you have saved a lot of money there. If you’re going to use a GEP for a tumor board discussion to de-escalate treatment, you can potentially save money there too.
BM: There was an abstract at the European Association of Dermato-Oncology (EADO) in Athens a year ago that if one uses GEP to decide who should be getting a sentinel lymph node biopsy, and that eliminates some of those borderline cases that have a good GEP, it is cost favorable. At least from that perspective, if we can take some of those people who fall into that “discuss and consider” range and shift them into the “don’t do it” range, there’s some tremendous cost savings there.
JV: Yes, I remember that paper; it was impressive. They took these patients who fell, by the Rule of Thumb, nomograms or whatever non-genetic tool was used to estimate risk of positive SLN into the “discuss and consider” range, 5% to 10% risk, and they ran a genetic expression profile. They were able to eliminate a lot of those patients from the NCCN “discuss and consider” no-man’s land; the GEP moved out approximately 60% of patients from “discuss and consider.” Most of them went into the “don’t do it,” less than 5% risk, category and about 12% went up to the “do it”, greater than 10% risk, category. If you can do anything to get patients out of that indecision land, that 5 to 10% NCCN limbo,
it’s beneficial.
DC: That’s great perspective on monetary resource utilization. With the little bit of time we have left, let’s talk about another important issue with resource utilization. We all know that tissue is always the issue. And when we’re doing these types of gene expression profile tests or ctDNA to generate the dataset that we’re going to use to either prognosticate or monitor our patients, we need genomic molecular material, which has to be extracted from tumor blocks. One fear that I have is burning through the block and not getting the important information you need, as it might relate to DecisionDx melanoma, the Merlin test, ctDNA, and/ or mutational analysis. How do you prioritize what molecular material you want? Because sometimes you can’t get it all.
JVA: Yeah, when I have a stage 3 or stage 4 melanoma, we have enough information to do next generation sequencing, look for BRAF, look for other potential mutations to help me predict what the response is going to be, or other potential treatments that I can offer my patients once immunotherapy doesn’t work. If it’s an advanced melanoma, stage 3, stage 4, because I know these patients are going to need treatment anyway, I prioritize the next generation sequencing, but when I don’t have tissue, I’m kind of stuck. I can do liquid biopsies in those cases. ctDNA is a good option. ctDNA in melanoma has a very high positive predictive value (85%–100%) for disease recurrence and is a strong indicator of treatment response or progression. Detectable ctDNA, especially postsurgery or during immunotherapy, indicates a high risk of relapse. However, I don’t know if we have really good data to help us escalate treatment in positive ctDNA patients, but we, in the community, should be open minded and incorporate all this information with caution to help us make decisions in specific cases.
BM: Practically, it has just not really been a big issue. I have quite a few patients where I order both GEP and ctDNA at baseline. There will be instances where whoever gets second bite at the apple has a technical failure. But it doesn’t happen very often, and I decide which to order first based on what is my main priority. Am I looking for a true risk stratification tool? Well, that’s GEP, then I’ll order that first. But if I have a really advanced melanoma, I may do ctDNA first. You can get ctDNA off any tissue: a biopsy, a wide local, or a metastatic foci. On the other hand, the GEP really just comes off either the biopsy or potentially the wide local. It just happens rarely that I don’t have enough.
DC: Thanks for that perspective. Dr. Moody. It’s important to hear that getting both hasn’t been a problem in your practice. Thanks for sharing your experience. Dr Vetto, did you have any additional thoughts?
JV: Maybe I’m just paranoid, but I do worry about block exhaustion, especially in these thinner melanomas. Let’s say you do a low-risk melanoma, but it’s node positive and now you want to order either a 3-gene mutation panel, or a Genetrails, or a Foundation test, whatever your institution orders, to determine the presence of a BRAF mutation and other drug targets. I often tell my staff that we have to go back to the block to get those tests, because you often can’t get that on the positive node, because the met in the node is often just a few cells. Additionally, the wide local excision specimen is often sterile; it contains no tumor. So, you often have to go back to the biopsy block to get these tests that are very important in patients who upstage or progress.
By the same token, I do agree with Dr. Moody that I do like, especially for the non-terrible, clinically early-stage melanomas, to get the GEP first. I think it is a good investment of tissue. It only requires
8 unstained slides, and I think that requirement is actually going down. Usually that test alone does not exhaust the block. Also, the drug target tests I mentioned can often be ordered later; if the patient progresses and the original block is exhausted all is not lost because you can still find the answers on a new biopsy.
DC: That’s awesome perspective. It was so cool hearing from each one of you and how you prioritize your molecular test for early stage vs late-stage disease. And it sounds like across the board, we’re all pretty much on the on the same wavelength there. I’ll just open it up as we close. Any last thoughts that you might have related to melanoma, molecular testing, thoughts for the readership, future directions, etc?
JV: I would just say to your readership that the NCCN guidelines are good. We all like them. But they are only guidelines, and they are subject to change, and they will change in regard to these GEPs. They are already changing. One thing to remember is that the NCCN consistently says that these tests should be studied in the setting of a clinical trial, and I completely agree. If you’re in your own setting and you want to use these tests, I’d strongly encourage you to order the test on a clinical trial, either an industry sponsored trial with the test you’re using, or an institutional trial, You can get your IRB to help you design a registry study so that we’re following the guidelines, but at the same time, we’re learning and we’re using tests that I think are an inevitable part of the future.
BM: I would like my dermatology colleagues to understand that our cancers, the ones that we’re primarily responsible for diagnosing and early management of, are being pulled into the modern cancer era. Other tumors have been a part of this world for a long time. We were sort of late to the game. Whether it’s solid tumors or hematologic malignancies, genetic profiles, mutational analysis, this is modern oncology, and to be in the modern oncology world, you need to be facile with these new tools to serve your patients in the best way possible.
JVA: My recommendation for my oncology colleagues is to be open minded about novel data and incorporate it into your pratice with caution for specific circumstances. There is more out there than just the NCCN guidelines. There are other things we can do for our patients. Scans are currently the standard of care, but there are other things that are going to come and they’re going to help us escalate and hopefully de-escalate, which is my ultimate goal for most of my melanoma patients, so they don’t have to experience the side effects of these immunotherapies.
DC: Wonderful. Thank you all for participating in this multidisciplinary melanoma roundtable focused on these complicated case discussions and the utility of molecular tools.
Conclusion
Melanoma remains the 5th most common cancer in the US and has a 2% death rate.1 Despite early detection, the fact that approximately 70% of CM cases are diagnosed as stage I, development of personalized molecular tools to prognosticate (GEP and ctDNA) and monitor therapeutic response (ctDNA), and incredible advancements in therapy, >8000 Americans die from metastatic melanoma annually.1–3 NCCN guidelines provide recommendations for the management of melanoma at the population level, and as such serve as guardrails for the average melanoma patient. However, a significant limitation is that the application of population-based medicine fails the individual when the individual lands outside of the population. Hence, the future of CM management requires integration with personalized tools to accurately predict risk of sentinel lymph node positivity, recurrence, distant metastasis, death, and response to therapy. Only in that way, can we safely and confidently deploy the appropriate healthcare resources, such as sentinel lymph node biopsy, surveillance imaging, adjuvant therapy, neo-adjuvant therapy, and other interventions on the horizon, to save the lives of >8,000 Americans that die from metastatic melanoma annually.
References
- Cancer Stat Facts: Melanoma of the Skin. Accessed 1 May 2026. https://seer.cancer.gov/statfacts/html/melan.html
- Keung EZ, Gershenwald JE. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: implications for melanoma treatment and care. Expert Rev Anticancer Ther. 2018;18(8):775–784.
- Helvind NM, Brinch-Møller Weitemeyer M, Chakera AH, et al. Stage-specific risk of recurrence and death from melanoma in Denmark, 2008-2021: a national observational cohort study of 25,720 patients with stage IA to IV melanoma. JAMA Dermatol. 2023;159(11):1213–1222.
- Sadrolashrafi K, Cotter DG. Not your mother’s melanoma: causes and effects of early melanoma diagnosis. Dermatopathology (Basel). 2022;9(4):368-378.
- Malvehy J, Hauschild A, Curiel-Lewandrowski C, et al. Clinical performance of the Nevisense system in cutaneous melanoma detection: an international, multicentre, prospective and blinded clinical trial on efficacy and safety. Br J Dermatol. 2014;171(5):1099–1107.
- Jarell A, Gastman BR, Dillon LD, et al. Optimizing treatment approaches for patients with cutaneous melanoma by integrating clinical and pathologic features with the 31-gene expression profile test. J Am Acad Dermatol. 2022;87(6):1312–1320.
- Podlipnik S, Martin BJ, Morgan-Linnell SK, et al. The 31-gene expression profile test outperforms AJCC in stratifying risk of recurrence in patients with stage I cutaneous melanoma. Cancers (Basel). 2024;16(2):287.
- Whitman ED, Koshenkov VP, Gastman BR, et al. Integrating 31-gene expression profiling with clinicopathologic features to optimize cutaneous melanoma sentinel lymph node metastasis prediction. JCO Precis Oncol. 2021;5:PO.21.00162.
- Guenther JM, Ward A, Martin BJ, et al. A prospective, multicenter analysis of the integrated 31-gene expression profile test for sentinel lymph node biopsy (i31-GEP for SLNB) test demonstrates reduced number of unnecessary SLNBs in patients with cutaneous melanoma. World J Surg Oncol. 2025;23(1):5.
- Kriza C, Martin B, Bailey CN, Bennett J. Integrating the melanoma 31-gene expression profile test with clinical and pathologic features can provide personalized precision estimates for sentinel lymph node positivity: an independent performance cohort. World J Surg Oncol. 2024;22(1):228.
- Glazer A, Tassavor M, Portela D, Soleymani T. The integrated 31-gene expression profile test (i31-GEP) for cutaneous melanoma outperforms the CP-GEP at identifying patients who can forego sentinel lymph node biopsy when applying NCCN guidelines. SKIN J Cutan Med. 2022;6:474–481.
- Yamamoto M, Sickle-Santanello B, Beard T, et al. The 31-gene expression profile test informs sentinel lymph node biopsy decisions in patients with cutaneous melanoma: results of a prospective, multicenter study. Curr Med Res Opin. 2023;39(3):417–423.
- Marchetti MA, Dusza SW, Bartlett EK. Utility of a model for predicting the risk of sentinel lymph node metastasis in patients with cutaneous melanoma. JAMA Dermatol. 2022;158(6):680–683.
- Hieken TJ, Egger ME, Angeles CV, et al. Gene expression profile–based test to predict melanoma sentinel node status: The MERLIN_001 Study. JAMA Surg. 2025;160(12):1358–1366.
- Beard T, Guenther JM, Leong SP, et al. The integrated 31-gene expression profile test identifies low-risk patients with cutaneous melanoma who can forego the SLNB procedure: results from a prospective, multicenter trial. Future Oncol. 2026;22(8):933–938.
- Ma VT, Zhou AY, Forati A, et al. Multi-institutional study evaluating the role of early circulating tumor DNA dynamics during treatment with immune checkpoint inhibitors in patients with advanced-stage melanoma. JCO Precis Oncol. 2026;10:e2500254.
- Ansstas G, Khaddour K, Sudhaman S, et al. Longitudinal ctDNA monitoring for postsurgical disease surveillance in patients with stage I to IIIB melanoma. Clin Cancer Res. 2026;32(8):1513–1521.
- Luke JJ, Rutkowski P, Queirolo P, et al; KEYNOTE-716 Investigators. Pembrolizumab versus placebo as adjuvant therapy in completely resected stage IIB or IIC melanoma (KEYNOTE-716): a randomised, double-blind, phase 3 trial. Lancet. 2022;399(10336):1718–1729.
- Mehta A, Motavaf M, Nebo I, et al. Advancements in melanoma treatment: a review of PD-1 Inhibitors, T-VEC, mRNA vaccines, and tumor-infiltrating lymphocyte therapy in an evolving landscape of immunotherapy. J Clin Med. 2025;14(4):1200.
- NCCN Clinical Practice Guidelines in Oncology. Melanoma: Cutaneous; Version 1.2026.
- Gannon CJ, Rousseau DL Jr, Ross MI, Gershenwald JE, et al. Accuracy of lymphatic mapping and sentinel lymph node biopsy after previous wide local excision in patients with primary melanoma. Cancer. 2006;107(11):2647–2652.
- Risk Calculators. Melanoma Institute of Australia. Accessed 1 May 2026. https://melanoma.org.au/for-clinicians/risk-calculators/
- Dhillon S, Duarte-Bateman D, Fowler G, et al. Routine imaging guided by a 31-gene expression profile assay results in earlier detection of melanoma with decreased metastatic tumor burden compared to patients without surveillance imaging studies. Arch Dermatol Res. 2023;315(8):2295–2302.
- Amaria RN, Reddy SM, Tawbi HA, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24(11):1649–1654.
- Han D, Han G, Duque MT, Vetto J, et al. Sentinel lymph node biopsy is prognostic in thickest melanoma cases and should be performed for thick melanomas. Ann Surg Oncol. 2021;28(2):1007–1016.
- Eroglu Z, Broman KK, Thompson JF, et al. Outcomes with adjuvant anti-PD-1 therapy in patients with sentinel lymph node-positive melanoma without completion lymph node dissection. Immunother Cancer. 2022;10(8):e004417.
- Bailey CN, Martin BJ, Petkov VI, et al. 31-gene expression profile testing in cutaneous melanoma and survival outcomes in a population-based analysis: a SEER collaboration. JCO Precis Oncol. 2023;7:e2300044.