by Elissa M. Schwartzfarb, BS, Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine, Miami, Florida;
Dot Weir, RN, CWON, CWS, Clinical Director, The Wound Center at Osceola Regional Medical Center, Kissimee, Florida;
Walter A. Conlan III, MD, CWS, Medical Director, The Wound Center at Osceola Regional Medical Center, Kissimee, Florida;
Paolo Romanelli, MD, Associate Clinical Professor, Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine, Miami, Florida;
Robert S. Kirsner, MD, PhD, Vice-Chairman and The Stiefel Laboratories Professor of Medical Dermatology, Department of Dermatology and Cutaneous Surgery, Department of Epidemiology and Public Health, University of Miami Miller School of Medicine, Miami, Florida
Abstract
Individuals with Bruton’s X-linked agammaglobulinemia (XLA) inherit a defect in the Btk gene, critical for B-cell differentiation. As a result, there is an absence of mature B-cells in the peripheral circulation with a marked reduction in serum levels of all immunoglobulin subtypes, predisposing patients with XLA to recurrent bacterial infections. Btk also functions in myeloid and dendritic cells, specifically in Toll-like receptor (TLR) signaling. TLRs are important in the recognition of foreign pathogens and elaboration of cytokines, such as tumor necrosis factor alpha (TNF-alpha). This suggests that the pathophysiology of XLA involves additional and unexplored immune dysregulation. The coexistence of pyoderma gangrenosum (PG) in a patient with Bruton’s XLA has been rarely reported. PG is an uncommon, ulcerating, neutrophilic dermatosis. Although its etiology is unknown, it is noninfectious and thought to involve abnormal immune and neutrophil responses. Anti-TNF agents have been effective in treating some patients with PG, suggesting TNF-alpha may play a role in the pathogenesis of PG. Here we report the association of PG and Bruton’s XLA, and demonstrate the presence of TNF-a within the lesion of PG.
(J Clin Aesthetic Derm. 2008;1(1):26–29)
*****************************************************************************************************************************************************
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency first characterized by Bruton in 1952.[1] Occurring in approximately 1 in 250,000 males, these individuals carry a mutation in the Btk gene encoding for a tyrosine kinase critical for B-cell maturation.[2] As a result, individuals with XLA have an absence of differentiated B cells and a decrease in all serum immunoglobulins. This defect in humoral immunity leads to increased susceptibility to infection, especially with encapsulated pyogenic organisms, such as Streptococcus pneumoniae, Haemophilus influenzae, and Pseudomonas species. Pneumonia, sinusitis, meningitis, and bacterial diarrhea are common, as is an increased susceptibility to enteroviral infections. Although no curative therapy exists, intravenous immunoglobulin (IVIg) is the mainstay of XLA treatment. Used for more than 20 years, IVIg has an excellent safety profile and has been shown to reduce morbidity and increase survival in patients with XLA.[3]
As impaired B-cell development is the most apparent phenotype of XLA, most work has focused on this cell type and clinical manifestations of low-serum immunoglobulin. Recent studies, however, suggest additional immune dysregulation may be involved in the pathogenesis of XLA. One report shows that stimulated T-helper cells from agammaglobulinemic individuals display a preferential Th1 profile.[4] Skewing of T-helper responses toward Th1 leads to a cytokine environment that favors macrophage activity and tumor necrosis factor alpha (TNF-alpha) production. Other molecular studies show that Btk, mutated in XLA, functions in lineages other than B cells. In myeloid and dendritic cells, Btk has been found to be a component of Toll-like receptor (TLR) signaling, important for recognition of foreign pathogens.[5] Activation of TLRs leads to production of cytokines, notably TNF-alpha, that contribute to the inflammatory response.[6]
The coexistence of pyoderma gangrenosum (PG) with Bruton’s XLA has been rarely reported, with only four reports in the literature.[7–10] PG is an uncommon inflammatory disorder of the skin characterized by papules or pustules that eventually erode to form deep ulcerations.[11] The lesions are painful with violaceous, undermined borders that often rapidly progress. In 50 percent of cases, PG is associated with an underlying systemic disease, including inflammatory bowel disease, rheumatoid arthritis, and myeloproliferative disorders.[12] Although the etiology is unknown, an immune-mediated process is implicated. Recently, the first randomized trial for treatment of PG demonstrated benefit with the use of the anti-TNF-alpha agent infliximab.[13] Other anti-TNF agents, such as etanercept, have also been anecdotally reported to be successful in the treatment of PG.[14] The success of TNF agents suggests that this proinflammatory cytokine may play a role in PG pathogenesis.
Here we report the coexistence of PG and Bruton’s XLA, and demonstrate, by immunohistochemistry, the presence of TNF-alpha with the lesion. Taking into account current research, the association of these diseases raises interesting questions on the pathophysiology of Bruton’s XLA.
Case Report
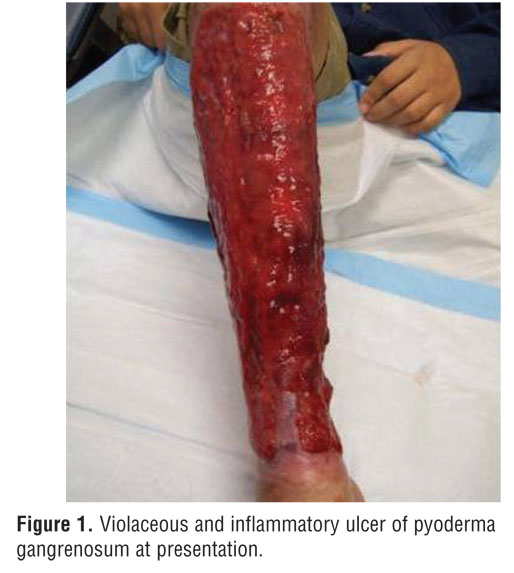
A 24-year-old man was referred to a University of Miami dermatology clinic for a large ulcerating leg lesion of four years duration (Figure 1). The lesion began as a few small ulcers on the right anterior leg that gradually coalesced and grew to circumferentially cover the entire right lower leg from ankle to knee. The patient’s medical history is significant for Bruton’s X-linked agammaglobulinemia. Diagnosed in infancy, he receives intravenous immunoglobulin replacement of 1g/kg/day every three weeks. Infectious complications encountered include recurrent sinusitis, one episode of infectious colitis three years prior, and meningitis one month prior. He has one affected brother and an affected maternal male cousin.
{kind=link}
On physical examination, the patient had a large 30- by 25-cm ulcer with raised violaceous borders covering his right lower leg. The base was erythematous and purple with granulation tissue present. He had no associated fever, malaise, or athralgias, and his pain was 10/10 with a burning sensation. Previous treatments included oral prednisone (1mg/kg/day) and dapsone (3mg/kg/day) without improvement. On two previous occasions, the lesion had been complicated by staphylococcal infection and was treated with vancomycin, levofloxacin, and trimethoprim/sulfamethoxazole.
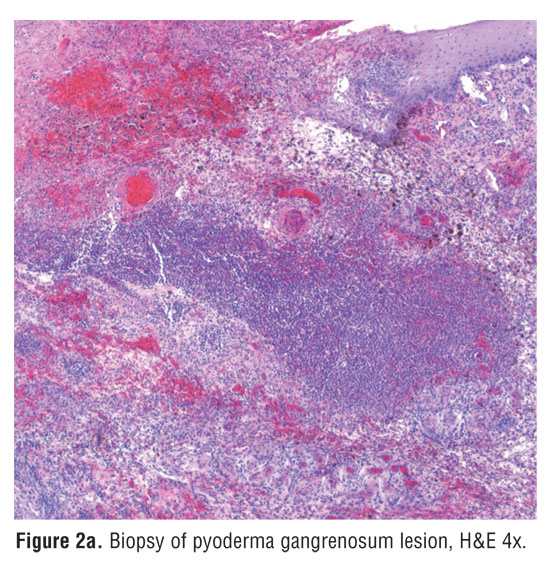
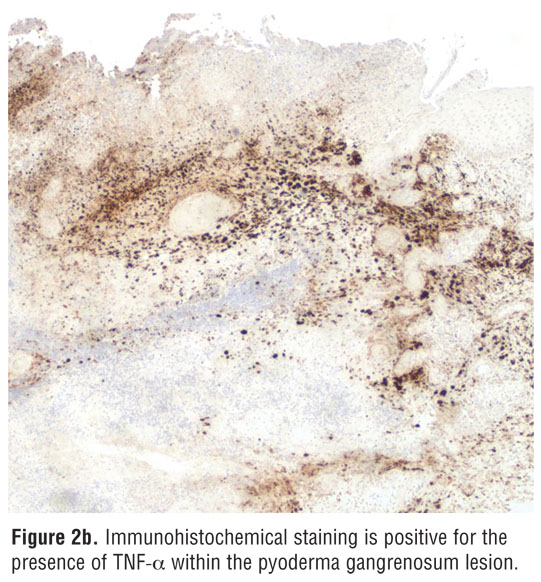
Histological evaluation of a biopsy specimen showed massive neutrophilic infiltration, hemorrhage, and necrosis of the overlying epidermis without findings of vasculitis. Immunohistochemical staining demonstrated the presence of TNF-alpha within the lesion, and its absence in unaffected areas (Figure 2a and Figure 2b).[15] Tissue cultures were negative for infectious organisms. Further laboratory data included a normal complete blood count with differential and serum chemistry panel. A decreased number of circulating B cells and low levels of all immunoglobulin subtypes were consistent with agammaglobulinemia (immunoglobulin A 16mg/dL [normal range 81–463mg/dL], immunoglobulin G 176mg/dL [normal range 694–1618mg/dL], immunoglobulin M <1mg/dL [normal range 48–271mg/dL]).
{kind=link}
{kind=link}
A diagnosis of PG was confirmed based on clinical presentation, exclusion of infectious origin, and histopathologic findings. The patient received treatment with four infusions of infliximab (5mg/kg/day over a three-month period), a TNF-alpha antagonist, with initial dramatic but not sustained improvement as assessed by recession of the borders and a cessation of the burning sensation. Current treatment includes doubling the patient’s dose of IVIg to 2g/kg/day given every three weeks in conjunction with prednisone 60mg/day. Follow-up at two months shows mild improvement of the ulcer as assessed by reepithelialization.
Discussion
Only four previous reports have documented the association of PG with XLA.[7–10] The most recently published report was a retrospective case series of 15 patients with XLA treated in a single tertiary care center.[7] In this large series, despite treatment with immunoglobulin substitution and antibiotics, infectious and noninfectious complications occurred. Two patients developed PG and were treated with both systemic steroids and IVIg. In one patient, the lesion cleared only after partial splenectomy for a splenic abscess.[7]
As XLA is a rare disease, a comprehensive clinical picture has been difficult to garner. Multiple case reports reveal autoimmune disorders in this population, specifically inflammatory bowel disease and rheumatoid arthritis.[16–20] Both of these diseases are characterized by increased levels of TNF-alpha and both are associated with the development of PG.[21–22]
Long understood to involve B-cell development, recent studies suggest that Bruton’s agammaglobulinemia involves additional and unexplored immune dysregulation, specifically the role of other cell lines and their mediators. Although a lack of antibodies in patients with XLA can account for an increase in bacterial infections, other immune alterations may account for the full clinical profile encountered in these patients. The Btk gene, known to be critical for B-cell differentiation, has also been found to function in myeloid and dendritic cells. Exactly what role it plays and how it translates to clinical practice remains unclear. A member of the Tec family of cytoplasmic tyrosine kinases, Btk appears to be a component of TLR signaling, important for sensing pathogens.[5] TLR activation leads to production of cytokines, notably TNF-alpha, which promote the inflammatory cascade.[6] Recent studies have attempted to determine if TNF-alpha production via TLRs is impaired in myeloid cells from XLA individuals. One group reports a decrease in TNF-a production by XLA monocytes when stimulated with LPS (a TLR-4 agonist), while another group reports the absence of any defect.[23–24] The former group also showed that monocytes, when differentiated toward macrophages, return to normal levels of TNF-alpha production, perhaps due to the accompanying increase in another Tec family kinase. Although results are uncertain, they suggest the pathophysiology of Bruton’s XLA may encompass more than simply a B-cell defect.
The pathophysiology of PG is also poorly understood. It is thought to involve abnormal neutrophil chemotaxis and alterations in cytokine levels.[25] Previously, ulcer specimens from PG lesions have been demonstrated to overexpress interleukin-8 (IL-8).[26] Here we show immunohistochemically the presence of TNF-alpha within a PG lesion. Both IL-8 and TNF-alpha are major chemotactic factors for neutrophils, in line with the observed neutrophilic infiltrate of PG lesions. The use of infliximab, a TNF-alpha antagonist, showed dramatic initial, but not sustained, improvement in our patient. This observation may be due to similar mechanisms found in patients treated for psoriasis, which have been attributed to rapid clearance of the drug either by antibody formation or other mechanisms.[14] Characteristically, no one treatment has been universally effective for PG, demonstrating the multifactorial nature of the lesion.
Conclusion
Taken together, the evidence suggests that the pathophysiology of Bruton’s XLA encompasses as of yet unexplored immune dysregulation. Further research can elucidate the pathways functioning in XLA and determine whether TNF-a or other cytokines play a role in the disease. The immune system, in light of decreased circulating antibodies, may favor intact immune defenses in Bruton’s agammaglobulinemia.
References
1. Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9:722–728.
2. Conley ME, Rohrer J, Minegishi Y. X-linked agammaglobulinemia. Clin Rev Allergy Immunol. 2000;19:183–204.
3. Quartier P, Debré M, De Blic J, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia. J Pediatr. 1999;134:589–596.
4. Amedei A, Romagnani C, Benagiano M, et al. Preferential Th1 profile of T-helper cell responses in X-linked (Bruton’s) agammaglobulinemia. Eur J Immunol. 2001;31:1927–1934.
5. Jefferies CA, Doyle S, Brunner C, et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem. 2003;278:26258–26264.
6. Kaisho T, Akira S. Toll-like receptor function and signaling. J Allergy Clin Immunol. 2006;117: 1191–1197.
7. van der Hilst JH, Smits BW, van der Meer JM. Hypogammaglobulinemia: cumulative experience in 49 patients in tertiary care institution. Neth J Med. 2002;60:140–147.
8. Barriere H, Litoux P, Stalder JF, et al. Pyoderma gangrenosum associated with congenital hypogammaglobulinemia. Ann Dermatol Venereol. 1979;106:695–696.
9. Bloom D, Fisher D, Dannenberg M. Pyoderma gangrenosum associated with hypogammaglobulinemia: report of two cases. AMA Arch Derm. 1958;77:412–421.
10. Marcussen PV. Hypogammaglobulinemia in pyoderma gangrenosum. J Invest Dermatol. 1955;24:275–280.
11. Callen JP. Pyoderma gangrenosum. Lancet. 1998;351:581–585.
12. Powell FC, Su WD, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395–409.
13. Brooklyn TN, Dunnill MGS, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double-blind, placebo-controlled trial. Gut. 2006;55:505–509.
14. Charles CA, Leon A, Banta M, Kirsner R. Etanercept for the treatment of refractory pyoderma gangrenosum: a brief series. Int J Dermatol. 2007;46:1095–1099.
15. Bebok Z, Szekeres G, Horvath G, et al. Creation of monoclonal antibodies against tumor necrosis factor-alpha and transforming growth factor alpha, their definition and possible use. Orv Hetil. 1993;134:1303–1307.
16. Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. QJM. 1993;86:31–42.
17. Washington K, Stenzel TT, Buckley RH, Gottfried MR. Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Am J Surg Pathol. 1996;20:1240–1252.
18. Verbruggen G, De Backer S, Deforce D, et al. X-linked agammaglobulinaemia and rheumatoid arthritis. Ann Rheum Dis. 2005;64:1075–1078.
19. Fu JL, Shyur SD, Lin HY, Lai YC. X-linked agammaglobulinemia presenting as juvenile chronic arthritis: report of one case. Acta Paediatr Taiwan. 1999;40:280–283.
20. Ersoy F, Sanal O, Tezcan I, Berkel AI. X-linked agammaglobulinemia: clinical and immunologic evaluation of six patients. Turk J Pediatr. 1990;32:241–247.
21. Stokkers PC, Camoglio L, van Deventer SJ. Tumor necrosis factor (TNF) in inflammatory bowel disease: gene polymorphisms, animal models, and potential for anti-TNF therapy. Journal of Inflammation. 1995–1996;47:97–103.
22. Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916.
23. Horwood NJ, Mahon T, McDaid JP, et al. Bruton’s tyrosine kinase is required for LPS-induced tumor necrosis factor alpha production. J Exp Med. 2003;197:1603–1611.
24. Impaired Toll-like receptor 8-mediated IL-6 and TNF-alpha production in antigen-presenting cells from patients with X-linked agammaglobulinemia. Blood. 2007;109:2553–2556.
25. Adachi Y, Kindzelskii AL, Cookingham G, et al. Aberrant neutrophil trafficking and metabolic oscillations in severe pyoderma gangrenosum. J Invest Dermatol. 1998;111:259–268.
26. Oka M, Berking C, Nesbit M, et al. Interleukin-8 overexpression is present in pyoderma gangrenosum ulcers and leads to ulcer formation in human skin xenografts. Lab Invest. 2000;80:595–604.