by Sanjay Bhambri, DO; Avani Bhambri, MD;
and James Q. Del Rosso, DO, FAOCD
Abstract
Calciphylaxis is a condition involving vascular calcification and cutaneous necrosis. The mortality rate is high, with more than 50 percent of patients dying within one year of diagnosis. The exact pathogenesis of calciphylaxis is unclear. Lower extremities are most commonly involved. Intravascular calcium deposition in the media of the dermal and subcutaneous arterioles is the diagnostic histopathologic feature seen. Treatment options include wound care, surgical debridement, sodium thiosulfate, bisphosphonates, and hyperbaric oxygen.
Introduction
Calciphylaxis, or calcific uremic arteriolopathy, is a rare condition, involving subcutaneous vascular calcification and cutaneous necrosis, which is seen most often in patients with renal failure. It was first described in 1898 by Bryant and White,[1] but it was not until 1962 that the term “calciphylaxis” was coined by Hans Selye.[2,3] The condition is also described in the literature as metastatic calcinosis cutis and necrotizing or calcifying panniculitis. It is important to note that the presence of renal failure is not an absolute requirement in patients with calciphylaxis, as calciphylaxis has been seen in the absence of renal failure. Diagnosis is made through a combination of clinical and histopathological features. There can be significant morbidity and mortality from the disease, most commonly resulting from septicemia due to impaired integrity of the epidermis and dermis.[4] More than 50 percent of patients die (most commonly from sepsis) within one year of being diagnosed.[5]
Pathogenesis
Calciphylaxis is a complex disorder with a multifactorial etiology. The exact pathogenesis of calciphylaxis is unclear. Medial calcification and intimal fibrosis of the cutaneous arterioles combined with thrombotic occlusion leading to ischemic skin necrosis is seen in calciphylaxis.
Calciphylaxis was defined as a disorder of hypersensitivity by Hans Selye.[2,3] In experimental animal models studied by Selye, administration of both a “sensitizer” and a “challenger” was required to elicit calcification in various organs. Either one alone was not sufficient in most cases. Excessive doses of “sensitizer” alone resulted in nonspecific metastatic calcification in predisposed tissues such as the cardiovascular and renal system. A critical period must elapse between sensitization and challenge in order for successful calcification to occur. A typical critical period was around 24 to 48 hours. Several sensitizers, such as dihydrotachysterol (DHT), vitamin D compounds, parathyroid hormone, and sodium acetylsulfathiazole (NaAST), have been identified. Among the challengers are salts of aluminum, lead, titanium, iron, and chromium; trauma; and organic compounds such as egg white and egg yolk.[6,7]
In humans, most of the cases are associated with chronic renal failure and end-stage renal disease (ESRD). Patients with ESRD usually have secondary hyperparathyroidism corresponding to increased levels of parathyroid hormone, calcium, phosphorous, and alkaline phosphatase. Secondary hyperparathyroidism, increased parathyroid levels, vitamin D, calcium, and phosphate may serve as sensitizers in humans, but identification of a specific challenger, such as corticosteroids, iron salts, and immunosuppressive drugs (such as methotrexate and cyclophosphamide), is imperative for successful therapy.[8] Similar sensitizers and challengers may be assumed to occur in patients with normal levels of parathyroid hormone or with normal renal function. Also, since most patients with ESRD and elevated calcium-phosphate product do not develop calciphylaxis, other mechanisms may be responsible for disease in those patients.
Most of the calcium in the calciphylaxis plaque is derived from preformed bone.[9] Calcium phosphate product (CPP) is measured by multiplying the total serum calcium by total serum phosphorus concentration. A CPP greater than 70mg2/dL2 may put patients at an increased risk for calcification but is not required for patients to develop calciphylaxis. Many patients with calciphylaxis have normal or low CPP.[5] Increased CPP also leads to matrix mineralization.[10]
Excess aluminum greater than 25ng/mL confers a four-fold increased risk and is thought to play a role in the pathogenesis of calciphylaxis.[5] Diabetes, obesity, corticosteroid use, immunosuppressive drugs, warfarin use, female sex, and protein C or S deficiency have been reported as risk factors for the development of calciphylaxis.[5,11,8,12]
Additionally, obesity confers a four-fold increased risk for calciphylaxis.[5] Chronic tension on the subcutaneous septa and arterioles, produced by excess adipose tissue, in combination with edema, may play a role in the increased number of proximally distributed calciphylaxis lesions seen in obese patients.[13]
In non-dialysis patients, prednisone use is usually seen in the majority of patients with calciphylaxis. Autoimmune disease or inflammatory disease, such as systemic lupus erythematosus (SLE), polymyositis, Sjogren’s, ulcerative colitis, and rheumatoid arthritis, may put patients at an increased risk for developing calciphylaxis.[5]
In humans, vascular calcification is an active process and is not sufficient to produce skin necrosis.[7] Vascular calcification and thrombosis are both required to produce lesions of calciphylaxis. Activation of nuclear factor kB (NFkB) is thought to be the final common pathway leading to vascular calcification.[7]
Clinical Features
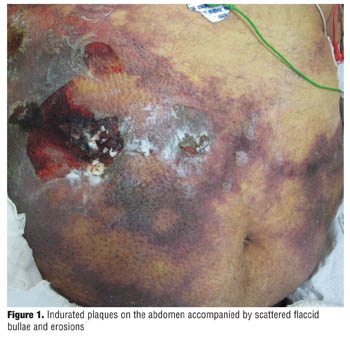
Calciphylaxis is more commonly seen in patients with ESRD, and females are affected more than males. Lower extremities are the most common area involved, with legs being the most common site. Lesions start with tender red areas developing into a livedoid pattern. Solitary or multiple indurated plaques and/or nodules are then seen. Patients may subsequently develop an eschar followed by frank ulceration, gangrene, or sepsis (Figure 1).[15]
Several patterns of involvement exist in patients with calciphylaxis. The distal pattern involves lower extremities while a proximal pattern involves the abdomen, inner thighs, and buttocks. Patients may have both proximal and distal patterns of involvement. Proximal involvement usually corresponds with a poorer prognosis. The survival rate was higher in patients who had distal localization of the disease (75.5% vs 26.2%).[5]
{kind=link}
Face and upper extremities are rarely involved. Patients may have palpable deposits of calcium,[16] and bullae may be noted. The most consistent feature of calciphylaxis is pain. Extreme pain is noted when skin around the ulcer is palpated.
Weenig et al[7] reviewed 64 patients with a diagnosis of calciphylaxis. They found that legs were the most common site (92%), followed by trunk (30%), buttocks, upper extremity, and genitalia. Of the 64 patients, 49 were on dialysis. Sixty-one percent of patients only had proximal lesions, 27 percent only had distal lesions, and 12 percent had both. Median survival was 2.64 months from the time of diagnosis. Sixty-seven percent of patients who died had cutaneous ulceration. Skin lesions included necrotic cutaneous ulcers, livedo racemosa (thrombotic vascular disorder of small blood vessels of the lower limbs), hemorrhagic patches, indurated plaques, and hemorrhagic bullae.
Histopathology
Intravascular calcium deposition in the media of the dermal and subcutaneous arterioles is the most common histopathologic feature seen in calciphylaxis.[14] Cutaneous necrosis may be seen at different levels. Vascular thrombosis of pannicular or dermal arterioles was noted in the majority of patients.[5,14] Extravascular calcification between and within lipocytes may also be seen.[14] Biopsy must include subcutaneous tissue for a proper diagnosis. Calcium deposition can be highlighted using the Von Kossa stain.
Management
Calciphylaxis has a dismal prognosis with up to 80-percent mortality.[16] A two-fold increase in mortality is seen when cutaneous ulcers develop.[17] The key is to prevent patients with known risk factors from developing calciphylaxis. For example, controlling blood sugars in a diabetic patient and monitoring calcium-phosphate homeostasis is imperative.
Wound care is of utmost importance and should include debridement of necrotic tissue periodically. Systemic antibiotics should be used, if indicated. Opioid pain medications should be used instead of morphine as byproducts of morphine can cause hypotension, thereby, slowing the flow in the pannicular arterioles, and thus increasing the risk of thrombosis.[16] Non-calcium, non-aluminum phosphate binder, such as sevelamer, can be used as an adjunctive therapy.
The role of parathyroidectomy in the treatment of calciphylaxis is controversial. It has been reported to be of some help in patients with primary hyperparathyroidism.18 Hafner et al19 noted a survival benefit in patients who underwent parathyroidectomy. Of 58 patients, 38 who underwent parathyroidectomy survived compared with 13 of 37 patients who did not undergo parathyroidectomy. Parathyroidectomy should be avoided in patients without proven primary hyperparathyroidism.[5]
Systemic corticosteroids are of no benefit and may exacerbate arteriolar calcification. Furthermore, corticosteroids may cause calcium and phosphorous abnormalities from adynamic bone disease.[5]
Cinacalcet is a calcimimetic that targets the calcium-sensing receptors of the parathyroid gland chief cells. It is used to lower parathyroid levels as well as improve calcium-phosphorus homeostasis. A dose of 30 to 60mg/d has been shown to be helpful in lowering parathyroid levels.[20]
Bisphosphonate therapy with etidronate disodium has been shown to be effective in treating patients with calciphylaxis. A possible mechanism may involve removing arterial calcification. A dose of 200mg/day for 14 days has been used with success, effectively lowering the calcium-phosphorus levels.[21]
Sodium thiosulfate (STS) has been used for many years for the treatment of cyanide and cisplatin intoxication. The half-life of STS in patients with normal renal function is 15 minutes. The exact mechanism of STS in calciphylaxis is unknown; however, it may play a role in chelating calcium from tissue deposits. A dose of 5 to 25g given intravenously, during or after hemodialysis, has been shown to be clinically effective.22 STS can cause acid-base imbalances, such as anion gap metabolic acidosis, which can be managed by using a high bicarbonate dialysate. Pain relief and reduction in skin lesions after a few weeks of using STS is usually seen. Araya et al[23] reported successful use of STS in three patients between 12 and 21 years of age. Time from ESRD to the diagnosis of calciphylaxis was 1, 9, and 20 years. STS dosage was 25g/1.73m2 per dose intravenously after each hemodialysis session. All patients showed rapid pain relief with improvement of skin induration and joint mobility of the extremities.
Hyperbaric oxygen therapy (HOT) consists of breathing 100-percent oxygen at higher than ambient pressure, with the patient inside a sealed chamber. The aim is to restore tissue Po2 to normal or above-normal levels and thus enhance angiogenesis, fibroblast proliferation, and collagen production.[24,25] HOT has been reported to be beneficial in the treatment of cutaneous ulcers in calciphylaxis.[24,25]
Summary
Calciphylaxis is a complex disorder with a multifactorial etiology. The exact cause is unknown, but it is seen more commonly in patients with renal failure. In those patients who have known risk factors, every effort must be taken to control or rectify them as the mortality rate is high. As our understanding into the pathogenesis improves, new insights will give rise to new therapies.
References
1. Bryant JH, White WH. A case of calcification of the arteries and obliterative endarteritis, associated with hydronephrosis, in a child aged six months. Guys Hosp Rep. 1898;55:17–20.
2. Selye H, Gabbiani G, Strebel R. Sensitization to calciphylaxis by endogenous parathyroid hormone. Endocrinology. 1962;71:554–558.
3. Selye H. The dermatologic implications of stress and calciphylaxis. J Invest Dermatol. 1962;39:259–275.
4. Oh DH, Eulau D, Tokugawa DA, et al. Five cases of calciphylaxis and a review of the literature. J Am Acad Dermatol. 1999;40:979–987.
5. Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56(4):569–579.
6. Vasku V, Vasku J. The development of the pathophysiological concept of calciphylaxis in experiment and clinic. Pathophysiology. 2001;7(4):231–244.
7. Weenig RH. Pathogenesis of calciphylaxis: Hans Selye to nuclear factor kappa-B. J Am Acad Dermatol. 2008;58(3):458–471.
8. Ozbalkan Z, Calguneri M, Onat AM, et al. Development of calciphylaxis after long-term steroid and methotroxate use in a patient with rheumatoid arthritis. Intern Med. 2005;44(11):1178–1181.
9. Lavender DR, Singer L, Armstrong WD. Nature and origin of the plaque produced in cutaneous calciphylaxis. J Dent Res. 1968;47(6):907–909.
10. Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol. 2004;15(12):2959–2964.
11. Goli AK, Goli SA, Shah LS, et al. Calciphylaxis: a rare association with alcoholic cirrhosis. Are deficiencies in protein C and S the cause? South Med J. 2005;98(7):736–739.
12. Conn J Jr, Krumlovsky FA, Del Greco F, et al. Calciphylaxis: etiology of progressive vascular calcification and gangrene? Ann Surg. 1973;177(2):206–210.
13. Janigan DT, Hirsch DJ. Does obesity play a role in the pathogenesis of calcific uraemic arteriolopathy? Nephrol Dial Transplant. 2006;21(4):865–868.
14. Au S, Crawford RI. Three-dimensional analysis of a calciphylaxis plaque: clues to pathogenesis. J Am Acad Dermatol. 2002;47(1):53–57.
15. Dosanjh A, Kebebew E. Calciphylaxis: rare but fatal. Curr Surg. 2005;62(5):455–458.
16. Rogers NM, Teubner DJ, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20(2):150–157.
17. Guldbakke KK, Khachemoune A. Calciphylaxis. Int J Dermatol. 2007;46(3):231–238.
18. Nigwekar SU. An unusual case of nonhealing leg ulcer in a diabetic patient. South Med J. 2007;100(8):851–852.
19. Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33(6):954–962.
20. Robinson MR, Augustine JJ, Korman NJ. Cinacalcet for the treatment of calciphylaxis. Arch Dermatol. 2007;143(2):152–154.
21. Hanafusa T, Yamaguchi Y, Tani M, et al. Intractable wounds caused by calcific uremic arteriolopathy treated with bisphosphonates. J Am Acad Dermatol. 2007;57(6):1021–1025.
22. Meissner M, Kaufmann R, Gille J. Sodium thiosulphate: a new way of treatment for calciphylaxis? Dermatology. 2007;214(4):278–282.
23. Araya CE, Fennell RS, Neiberger RE, et al. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin J Am Soc Nephrol. 2006;1(6):1161–1166.
24. Basile C, Montanaro A, Masi M, et al. Hyperbaric oxygen therapy for calcific uremic arteriolopathy: a case series. J Nephrol. 2002;15(6):676–680.
25. Basile C. Hyperbaric oxygen therapy in the treatment of calciphylaxis? Yes, more than hope. Kidney Int. 2002;62(6):2300.