 J Clin Aesthet Dermatol. 2018;11(10):17–23
J Clin Aesthet Dermatol. 2018;11(10):17–23
by Caroline Vinkel, MD and Simon Francis Thomsen, MD, PhD
Drs. Vinkel and Thomsen are with the Department of Dermatology at Bispebjerg Hospital in Copenhagen, Denmark. Dr. Thomsen is also with the Department of Biomedical Sciences at the University of Copenhagen in Copenhagen, Denmark.
FUNDING: No funding was provided for this study.
DISCLOSURES: The authors have no conflicts of interest relevant to the content of this article.
ABSTRACT: Hidradenitis suppurativa (HS), a relatively common and chronic inflammatory skin disorder. HS can have debilitating consequences if not diagnosed and treated appropriately. Clinically defined by recurrent, inflamed nodules in intertriginous regions (i.e., axillary, inguinal, and perianal areas), HS can cause intense pain and, in severe disease stages, lead to the formation of fistulas, sinus tracts, and extensive scarring. Postpubertal onset and female preponderance further characterize HS. Numerous pathogenic mechanisms have been proposed in HS, including immune dysregulation, genetics, smoking, and obesity; however, the exact etiology remains to be elucidated. The association of HS with inflammatory bowel disease, cardiovascular disease risk factors, and psychiatric disorders suggests HS is a systemic disease. HS significantly impairs quality of life in patients in excess versus other skin diseases. Unfortunately, experiences indicate long diagnostic delays, which in many cases might be due to disease unawareness among physicians. Increased knowledge of HS is therefore important in order to optimize disease management and ultimately improve the quality of life of patients.
KEYWORDS: Hidradenitis suppurativa, inflammatory skin disease
Hidradenitis suppurativa (HS) is a chronic or relapsing inflammatory skin disease affecting hair follicles in intertriginous areas, including the axillary, inframammary, inguinal, genital, buttocks, and perianal/perineal areas.1 With a prevalence of 1 to 4 percent, HS is not an uncommon disease.2 It affects three times as many women as men, and one-third of HS patients report a positive family history. Other predisposing factors include smoking and obesity.3 The clinical features of HS are recurrent, painful nodules and abscesses, fistulas, sinus tracts (tunnels), and scarring. Patients with HS have significantly impaired quality of life due to both physical and psychological discomfort associated with the disease (e.g., pain, suppuration, social isolation, and work disability).4–6 Several conditions, including inflammatory bowel disease, metabolic syndrome, arthritis, and depression, have been reported to co-occur with HS.3
Early diagnosis and treatment is essential to prevent debilitating consequences of HS and improve quality of life in patients. Even though the knowledge of HS has increased tremendously in recent years, clinical experiences still indicate under-recognition and significant diagnostic delay of HS, which ultimately prevents many patients from receiving adequate treatment in time.7
HS is commonly encountered by nondermatologists (e.g., general practitioners, surgeons, gynecologists); hence, these individuals play a pivotal role in early diagnosis, initial treatment, and referral of patients to a dermatologist. Thus, widespread knowledge of clinical features and management methods of HS is essential in the primary care setting and other medical specialties.
This review provides information about the etiology, pathogenesis, clinical features, and current therapeutic options of HS based on the available literature.
Epidemiology
HS has been an overlooked disease for decades, and as a result, epidemiologic data are sparse. Reported prevalence rates for HS range from 0.03 to 4.1 percent depending on the studied population and study design.2 Although broadly accepted to be one percent in the general population, the true prevalence might be higher due to continued undiagnosis and misdiagnosis of patients. Recent data suggest increasing incidence of HS, as the incidence has more than doubled from 4.0 patients per 100,000 in 1968 to 10.0 patients per 100,000 in 2008.8 This tendency is most likely reflects improved diagnostics of patients due to increased awareness of the disease.
The average onset of HS is in the early 20s, and substantial data suggest a female-predominance with a 3:1 sex ratio.9,10 Earlier disease debut is reported in patients with family history of HS.10 HS has a prolonged course, with intermittent periods of activity and remission. Inflammatory activity usually peaks in the third and fourth decades of life and afterwards tends to resolve, especially in women after menopause.10 Most patients have mild or moderate disease, though severe disease has been reported in 4 to 22 percent of patients.9,11 An average diagnostic delay of seven years has been reported among HS patients in specialized clinics; however, even longer delays can be experienced by patients outside of such clinics.7
Etiology, Pathogenesis, and Risk Factors
Pathogenic mechanisms. The pathogenesis of HS is not entirely established. Current knowledge no longer supports the original theory of HS as an infectious disease of apocrine sweat glands (hidros meaning “sweat” and aden meaning “gland”). Newer research suggests HS to be an inflammatory disease with dysregulated skin immunity around hair follicles in intertriginous regions.10,12–14 Based on a suggested shared pathogenesis involving follicular occlusion, HS has been associated with severe acne (acne conglobata), dissecting cellulitis of the scalp, and pilonidal disease under the term “the follicular occlusion tetrad.”15 Initial pathogenic events in HS involve perifollicular inflammation leading to hyperkeratosis and occlusion of hair follicles in the predilection areas. Subsequent rupture of the dilated follicle and extrusion of accumulated follicular duct content (sebum and debris) into the surrounding dermis initiates an inflammatory response, ultimately resulting in the formation of painful, inflamed nodules. Sustained inflammation in the area contributes to the formation of abscesses, fistulas, and sinus tracts (tunnels), thus creating a favorable environment for biofilm formation, bacterial colonization, and suppuration.13
Dysregulated immunity. The association of HS with other well-established inflammatory diseases, such as Crohn’s disease (CD) and pyoderma gangrenosum (PG), along with the beneficial effects of antitumor necrosis factor (TNF)-agents suggests a primary role of dysregulated immunity in HS pathogenesis.13,16 Furthermore, several studies have demonstrated elevated levels of pro- and anti-inflammatory cytokines, most notably interleukin (IL)-1beta, TNF, IL-17, and IL-10 in lesional skin, as well as abnormalities in antimicrobial peptides and Toll-like-receptor signaling.17,18 The IL-23/TH17 pathway is suggested to be involved in HS and, especially, the role of IL-17; IL-17 is an essential factor in other inflammatory diseases, including psoriasis and CD.19 However, detailed cytokine profiles and exact pathogenic pathways in HS need further elaboration.
Bacteria. Bacterial involvement in HS pathogenesis remains highly debated.20 Cultures of HS lesions are predominately sterile or show commensal skin flora.1 Hence, primary infection is considered an unlikely causative factor in HS.1 However, a variety of species have been isolated from HS lesions and alterations in the skin microbiome is a possible contributing factor in HS pathogenesis.21,22 Fistulas and sinus tracts provide an opportunity for biofilm formation, bacterial colonization, and secondary infection, which contributes to disease exacerbations, suppuration, and extension of lesions.10
Genetics. One-third of patients with HS report a positive family history, indicating a genetic pattern in HS.23 Genetic mutations in gamma-secretase genes, supposedly leading to abnormal epidermal proliferation and differentiation, have been identified in a minority of patients with HS.24 Several additional genetic alterations have been linked to HS, suggesting genetic heterogeneity in HS.23,25 The contribution of genetics in HS pathogenesis remains unclear.
Smoking. More than 70 percent of patients with HS are smokers, and a strong association between smoking and HS has been demonstrated.8,26 The suggested pathogenic mechanism of smoking is through the stimulation of epidermal hyperplasia and inflammation.16 However, studies investigating the dose-response relationship between smoking and disease severity report conflicting results.8,11,26 Moreover, the temporal relationship between smoking and HS, as well as the effects of smoking cessation on disease course, need further elaboration.
Obesity. Obesity is a frequently proposed risk factor for HS. The prevalence of obesity is significantly higher in patients with HS as compared with healthy controls.27 Several studies report an association between body mass index (BMI) and HS and disease severity.1,9,11,26 Adiposity supposedly contributes to HS pathogenesis by stimulating a low-grade proinflammatory environment and by increasing mechanical stress (i.e., friction) at HS sites, which in turn promotes follicular hyperkeratinization and occlusion.13,16,27 There is increasing evidence of improvements in HS occurring following weight-reduction in patients with obesity.28
Clinical Features
The diagnosis of HS relies entirely on the clinical features and requires the following three criteria: 1) typical morphology (nodules, abscesses, sinus tracts, scars); 2) characteristic distribution or typography of lesions (intertriginous areas, axillae, inframammary folds, groins, buttocks, perianal and perineal areas); and 3) a relapsing, chronic disease course.29
The prototypic patient with HS presents with recurrent, painful, inflamed nodules, most commonly in the axillae and/or inguinal areas. However, there is substantial heterogeneity in the clinical features of HS. Gluteal, perianal/perineal, and atypical areas (e.g., ears, chest, and legs) are more frequently affected in male patients, while frontal areas (inguinal and inframammary folds) are more affected in female patients.8,9 Furthermore, increased disease severity is associated with higher BMI, atypical lesions, and severe acne.9 Classification of HS into the following three subtypes have been proposed using latent class analysis30: axillary-mammary (48%), follicular (26%), and gluteal (26%). In contrast to the axillary-mammary subgroup, the follicular subtype is characterized by atypical and follicular lesions (e.g., pilonidal sinus, comedones, severe acne). Furthermore, the follicular subgroup is associated with male sex, current smoking, positive family history of HS, earlier disease onset, and increased disease severity. Besides gluteal involvement, patients in the gluteal subgroup typically have lower BMIs and a less severe disease course.
Autoinflammatory syndromes caused by specific genetic mutations in the inflammasome associated with HS have been reported recently in the literature, including PASH (PG, acne, and HS) and PAPASH (pyogenic arthritis, PG, acne, and HS) syndromes.31,32 Syndromic HS is characterized by a more severe HS phenotype coexisting with severe acne, PG, and/or arthritis, accompanied by recurrent episodes of fever and increased inflammation markers in laboratory findings.33
Disease Assessment
Several clinical scoring systems to assess disease severity of HS exist. The most commonly used is the Hurley staging system, in which the mild disease (Hurley stage I) is characterized by single or multiple nodules and/or abscesses without permanent lesions; moderate disease (Hurley stage II) is defined by widely separated recurrent nodules and/or abscesses with sinus tracts and scarring; and severe disease (Hurley stage III) is determined by multiple interconnected sinus tracts, abscesses, and scarring affecting the entire anatomical area.1 The Hurley system is useful in daily clinical practice for overall disease classification, but is not dynamic nor sufficient for monitoring medical treatment response. Conversely, the modified Sartorius score represents a more dynamic and quantitative severity assessment tool (based on lesion count, number of anatomical regions involved, lesion type, and distance between two relevant lesions).10,11 It is, however, time consuming to use, difficult to interpret, and has limited applicability in severe disease stages where lesions are confluent. In addition, the HS Physician’s Global Assessment (PGA) and HS Clinical Response (HiSCR) scoring system are two validated and easy-to-use options mainly used in clinical trials and research.34
The use of ultrasound, particularly color Doppler, in HS severity assessment is increasing. There is emerging evidence that clinical examination alone can underestimate disease severity. Ultrasound allows for the visualization of subclinical changes (e.g., dermal thickening, dermal pseudocysts, widening of hair follicles, fluid collections) along with detailed information of lesion type and extent. Hence, ultrasound should be incorporated in diagnosis and assessment of disease severity.35,36
Besides the assessment of disease severity, patient-reported outcomes (e.g., Dermatology Life Quality Index [DLQI] and visual pain analog scale) and the evaluation of cardiovascular disease risk factors (e.g., obesity, smoking, metabolic syndrome) as well as other associated conditions should be included in the monitoring of patients.1,4,37,38
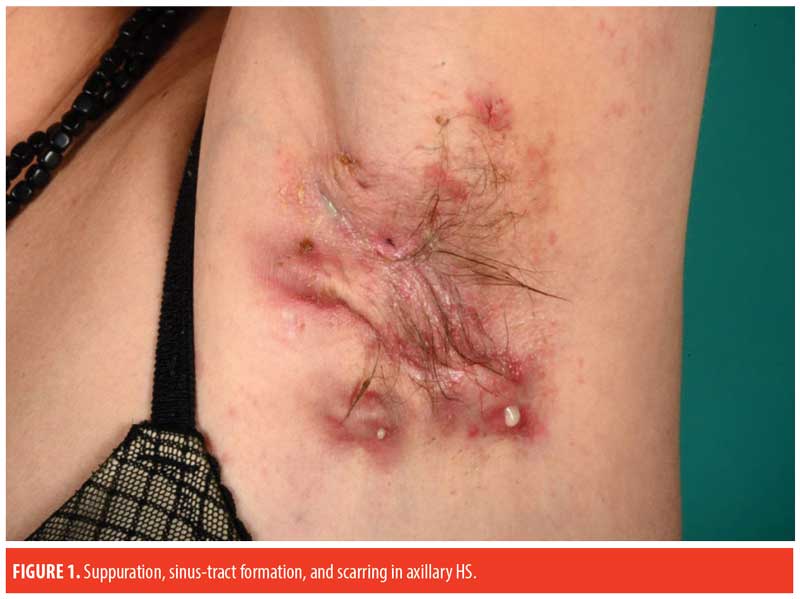

Paraclinical Examination and Findings
Since HS diagnosis is based exclusively on clinical features, there are no definite paraclinical findings.
Patients are usually afebrile and unaffected. Bacterial cultures are predominately sterile, and skin biopsy-histopathology is nonspecific. Routine blood tests including complete blood cell count with differential and platelet counts, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) are usually within normal limits; however, inflammatory markers (i.e., leukocytes, ESR, CRP) can be elevated in periods of active inflammation or in patients with more severe disease. Further relevant examinations in patients with HS include the assessment of BMI and blood pressure and the measuring of blood glucose and lipid levels.34,38,39
Differential Diagnosis
The characteristic distribution of lesions and postpubertal onset helps distinguish HS from other conditions with similar morphology, such as nodular acne. Microbiology cultures can be used to differentiate HS from infectious entities (sterile or mixed growth vs. growth of single infectious agents) such as abscesses, carbuncles, and furuncles.10
Cutaneous manifestations of CD can be difficult to separate from perianal HS lesions. Perianal lesions concomitant with gastrointestinal symptoms favor CD, whereas involvement of the axillae or other HS-specific areas is suggestive of HS. Furthermore, ulcerative lesions and formation of endoanal fistulas possibly involving the anal sphincter are characteristic for CD and not HS.10,40
Malignancies (primary or metastatic) are usually accompanied by systemic signs of cancer (e.g., unexplained weight loss, fatigue, fever).
Similarly to HS, Hailey-Hailey disease, also known as benign familial pemphigus, involves painful, recurrent lesions in intertriginous areas. However, Hailey-Hailey disease is clinically characterized by erythematous vesicular plaques that can become crusted and develop into painful cracks.41
Comorbid Diseases
Several possible comorbidities have been associated with HS, most notably inflammatory bowel disease, cardiovascular disease, and psychiatric diseases.
HS has been reported in up to 26 percent of patients with CD42 and several similarities exist between the two. Both are chronic inflammatory diseases characterized by compromised barriers (intestinal and cutaneous) and abnormal immune response to commensal bacteria. Furthermore, in both conditions, responses to anti-TNF therapy are beneficial, smoking and genetic predisposition are considered risk factors, and fistula formation is a characteristic disease feature.43
Although rare, the coexistence of HS and PG has been reported in the literature.44 A shared pathogenesis involving cytokine dysregulation has been suggested. Several syndromes associated with PG and HS have been named, including the aforementioned PASH and PAPASH syndromes, supplied by PASS (PG, acne conglobata, HS, and axial spondyloarthopathy) syndrome.31–33,45
Increasing evidence proposes higher prevalence rates of cardiovascular disease risk factors, including metabolic syndrome, atherosclerosis, obesity, and smoking in HS patients versus in healthy controls.27,37,38,46 The increased systemic inflammatory load in HS has been suggested to be a contributing factor to the development of insulin resistance, dyslipidemia, endothelial dysfunction, and atherosclerosis.3
A high incidence of depression among HS patients is supported by substantial literature.5,8,47–49 It has been postulated that HS patients suffer from more severe depression as compared with in the case of other skin diseases.5,49 Additionally, there seems to be an association between HS severity, inflammation, and depression.49 Associations between HS and other psychiatric disorders (e.g., anxiety, schizophrenia, psychoses, and bipolar disorder) have also been observed.48
Most studies investigating possible comorbidities in HS are observational and therefore cannot prove causality. Nevertheless, the emerging evidence of systemic manifestations coexisting with HS raises the question of whether HS is a systemic disease.
Complications
Both physical and psychological complications can result from long-term, unmanaged disease. HS lesions can become secondarily infected. Erysipelas and sepsis following soft tissue infection have been reported in rare cases. Impaired joint mobility due to extensive fibrosis and scarring can occur, especially in the axillary region. Lymphedema and squamous cell carcinoma can develop in areas of chronic inflammation. Additional complications resulting from prolonged systemic inflammation include anemia, hypoalbuminemia, and AA amyloidosis leading to kidney failure.10,34
The intense pain, malodorous discharge, and disfiguring lesions in intimate places make HS a burdensome disease. The psychosocial sequelae accompanying HS, such as embarrassment, self-consciousness, social isolation, stigmatization, depression, sexual dysfunction, and work disability contributes to increased sick leave and a significant quality of life impairment which exceeds that of other disabling skin disorders.4–6
Treatment
Treatment of HS is challenging due to the lack of pathophysiologic insight and the recurrent nature of the disease. There are no curative (medical) therapies for HS, but symptoms can be managed effectively. Treatment guidelines have been developed in recent years. The current algorithm of the European Academy of Dermatology and Venereology and the European Dermatology Forum (S1 guidelines) recommend a multifaceted approach in the treatment of HS, ranging from adjuvant therapy (e.g., pain management, smoking cessation, weight loss, treatment of superinfections, hygiene practices, topical wound dressings) to topical and systemic agents (e.g., antibiotics, anti-inflammatory agents, biologics) and surgical interventions such as excisional surgery and laser surgery.1 The choice of treatment depends on the number, type, distribution, and anatomic location of lesions, as well as associated risk factors or comorbid diseases. Many of the suggested treatment interventions for HS rely on low-quality evidence due to the existence of only a few small and mostly uncontrolled studies. Furthermore, there is considerable variability in outcome measures, which makes comparison between the available studies difficult.50,51
Topical treatment. Topical clindamycin (1%) has been tested in a small randomized controlled trial (RCT) of 30 patients with mild localized disease (Hurley stages I and II). Significant clinical improvement was obtained after 12 weeks of treatment with topical clindamycin as compared with placebo.52 Application of clindamycin twice daily for one week is recommended to manage disease flare-ups in patients with mild localized disease. Long-term treatment with topical antibiotics should be avoided due to the risk of antimicrobial resistance.53 In the case of acutely inflamed nodules, intralesional glucocorticoid injections (triamcinolone 10mg/mL) or incision and drainage can yield immediate relief.54 Azelaic acid or topical retinoids have not been tested to date in any RCTs, but collective clinical experience suggests a beneficial effect of daily application and they are recommended as long-term treatment.1
Systemic treatment. Patients with more widespread disease and/or severe disease (Hurley stages II and III) further require systemic therapy. Systemic treatment regimens include antibiotics (e.g., tetracycline, rifampicin+clindamycin) and anti-inflammatory agents (e.g., anti-TNF agents, ciclosporin). The rationale for antimicrobial therapy in HS is not well established; however, it is mainly believed to rely on the anti-inflammatory effect of the drugs. Surgical intervention should always be considered alongside medical treatment to obtain complete remission of persistent lesions. Systemic tetracycline 500mg twice daily for at least three months is recommended in mild to moderate widespread disease in which topical treatment alone is inadequate.1 There is no sufficient high-quality evidence favoring systemic tetracycline over topical treatment. In fact, a small RCT of 46 patients with mild HS (Hurley stages I and II) showed no significant difference between systemic tetracycline and topical clindamycin, thus questioning the rationale for systemic tetracycline.55 However, systemic tetracycline is often preferred in patients with more widespread disease because it is easier for the patient to manage. When choosing between topical or systemic treatment in a patient with mild-to-moderate HS, it is important to consider the distribution of lesions, the extent of the disease, and individual patient factors (e.g., patient preferences, comorbidities, pregnancy). The combined use of systemic rifampicin (300mg twice daily) and clindamycin (300mg twice daily) is recommended following a lack of response to systemic tetracycline and/or in more advanced cases. Significant improvement in both clinical features and quality of life upon use of this regimen have been demonstrated in three open case series,56–58 but no prospective RCTs have yet been conducted to confirm these results. Systemic treatments with anti-inflammatory or immunomodulatory agents such as acitretin, isotretinoin, dapsone, ciclosporine, prednisone, and colchicine have been suggested. However, their efficacy has only been evaluated in case reports and small case series with varying results, so they are not recommended as standard regimens and should only be used in cases of inadequacy, intolerance, and/or contraindication (e.g. comorbidities, pregnancy) to the abovementioned therapies.1 Biologics (e.g., adalimumab [anti-TNF] or infliximab [anti-TNF]) are indicated in moderate-to-severe HS (Hurley stages II and III) recalcitrant to previous treatments.1 In two phase III RCTs that included 307 and 327 patients, respectively, with moderate-to-severe HS, adalimumab 40mg taken weekly prompted significant clinical improvement (?50% reduction in the total count of inflamed nodules/abscesses and no increase in abscess or draining-fistula counts, as well as a significant reduction in pain and DLQI score) after 12 weeks versus placebo.59 Adalimumab 40mg twice weekly is, however, ineffective.51 Evaluation of infliximab 5mg/kg in an RCT of 38 patients with moderate-to-severe HS showed no significant effect on the primary endpoint (>50% improvement) compared to placebo; however, a significantly higher 25- to 50-percent improvement rate was detected, as was a decrease in DLQI and VAS pain scores. Hence, infliximab is recommended secondarily to adalimumab.50,60 The efficacies of etanercept (anti-TNF) and anakinra (anti-IL1) have been investigated in case reports and small RCTs.1,61 No significant difference was found between etanercept 50mg weekly and placebo, whereas treatment with anakinra yielded significant improvement. Studies evaluating the efficacy of drugs targeting IL-12/23 as well as IL-17 are limited but highly expected in the future as these drugs become more accessible.62–64
Surgical treatment. Persistent lesions and diffuse scarring in HS are difficult to manage with medical treatment alone and often require surgery. To obtain the best results, surgery should be carried out during a period of minimum inflammatory activity in the patient’s HS. Intensified medical treatment can be necessary prior to surgery. Surgical procedures to manage HS include local and wide excisions, incision and drainage, deroofing, and laser techniques (e.g., carbon dioxide, neodymium-doped yttrium aluminum garnet [Nd:YAG]). Options for postoperative wound healing include primary closure, secondary intention wound healing, or various reconstruction techniques such as skin grafting or skin flaps. The type of surgical method and the pathway of healing depend on various factors, such as the localization and size of the lesions, as well as patient-related factors. As with all surgical procedures, postoperative complications include bleeding, infection, nerve damage, and stricture due to scar tissue. There are no studies providing guidance for the most optimal surgical technique or timing for when to perform surgery in HS.51 Further larger and well-executed studies are needed to optimize surgical treatment strategies for HS.
Incision and drainage is one of the most frequently performed procedures. However, the procedure is associated with high recurrence rates and should be restricted to use for the management of acutely inflamed abscesses.65
Excisions represent a meaningful procedure in otherwise well-managed HS in patients with individual, persisting lesions. Minor excisions are associated with a high risk of recurrence, whereas wide excisions have the lowest rates of recurrence.1 However, removal of entire areas can be extensive and require high surgical expertise (general or plastic surgeon) and reconstructive techniques.
Deroofing represents a technique in which the “roof” of the lesion is removed and the wound is left open for secondary intention healing. The procedure is especially suitable for the management of sinus tracts.
CO? laser evaporation or excision with secondary intention healing, and Nd:YAG laser are considered minimally invasive and tissue-saving procedures; however, further information regarding safety of the procedures, efficacy, and recurrence rates is warranted.1
Adjuvant therapy, such as the management of pain, treatment of superinfections, smoking cessation, weight reduction, and the mitigation of psychological sequelae and comorbid conditions should be considered in all patients with HS.1
Conclusion
HS is a relatively common and disabling disease that greatly affects the lives of those with the condition. Early diagnosis and treatment are essential to prevent morbidity (physical and psychological) related to untreated disease and to improve quality of life in patients. Thus, heightened awareness of the associated clinical features (i.e., morphology, typography, and disease course), risk factors (e.g., smoking, obesity, and family history), and possible comorbid conditions (e.g., irritable bowel syndrome, cardiovascular risk factors, and psychiatric disorders) among physicians encountering patients with HS is important to ensure timely referral to a dermatologist. Collective experiences so far suggest a multifaceted treatment approach that includes anti-inflammatory medicaments and surgical methods is best. However, the unknown etiology and recurrent nature of HS complicates efficient disease management. Improved targeted therapies rely on the continuous elaboration and investigation of pathogenic mechanisms implicated in HS, including dysregulated immunity, skin microbiome, and genetics. Furthermore, studies evaluating the efficacy of current and future treatment modalities are required to further substantiate evidence-based guidelines.
References
- Zouboulis CC, Desai N, Emtestam L, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29(4):619–444.
- Jemec GBE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(5 Suppl 1):S4–S7.
- Miller IM, McAndrew RJ, Hamzavi I. Prevalence, risk factors, and comorbidities of hidradenitis suppurativa. Dermatol Clin. 2016;34(1):7–16.
- Esmann S, Jemec GBE. Psychosocial impact of hidradenitis suppurativa: a qualitative study. Acta Derm Venereol. 2011;91(3):328–332.
- Matusiak L, Bieniek A, Szepietowski JC. Psychophysical aspects of hidradenitis suppurativa. Acta Derm Venereol. 2010;90(3):264–268.
- Matusiak ?, Bieniek A, Szepietowski JC. Hidradenitis suppurativa markedly decreases quality of life and professional activity. J Am Acad Dermatol. 2010;62(4):706–708.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173(6):1546–1549.
- Vazquez BG, Alikhan A, Weaver AL, et al. Incidence of hidradenitis suppurativa and associated factors: a population-based study of Olmsted County, Minnesota. J Invest Dermatol. 2013;133(1):97–103.
- Canoui-Poitrine F, Revuz JE, Wolkenstein P, et al. Clinical characteristics of a series of 302 French patients with hidradenitis suppurativa, with an analysis of factors associated with disease severity. J Am Acad Dermatol. 2009;61(1):51–57.
- Revuz J. Hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2009;23(9):985–998.
- Sartorius K, Emtestam L, Jemec GBE, Lapins J. Objective scoring of hidradenitis suppurativa reflecting the role of tobacco smoking and obesity. Br J Dermatol. 2009;161(4):831–839.
- Kurzen H, Kurokawa I, Jemec GBE, et al. What causes hidradenitis suppurativa?. Exp Dermatol. 2008;17(5):455-6; discussion 457–472.
- Yazdanyar S, Jemec GBE. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24(2):118–123.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122(6):763–769.
- Vasanth V, Chandrashekar BS. Follicular occlusion tetrad. Indian Dermatol Online J. 2014;5(4):491–493.
- Kelly G, Prens EP. Inflammatory mechanisms in hidradenitis suppurativa. Dermatol Clin. 2016;34(1):51–58.
- Kelly G, Hughes R, McGarry T, et al. Dysregulated cytokine expression in lesional and nonlesional skin in hidradenitis suppurativa. Br J Dermatol. 2015;173(6):1431–1439.
- Kelly G, Sweeney CM, Tobin A-M, Kirby B. Hidradenitis suppurativa: the role of immune dysregulation. Int J Dermatol. 2014;53(10):1186–1196.
- Kelly G, Prens EP. Inflammatory mechanisms in hidradenitis suppurativa. Dermatol Clin. 2016;34(1):51–58.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24(10):727–731.
- Ring H, Bay L, Kallenbach K, et al. Normal Skin Microbiota is Altered in Pre-clinical Hidradenitis Suppurativa. Acta Derm Venereol. 2017;97(2):208–213.
- Guet-Revillet H, Jais JP, Ungeheuer MN, et al. The microbiological landscape of anaerobic infections in hidradenitis suppurativa: a prospective metagenomic study. Clin Infect Dis. 2017;65(2):282–291.
- Ingram JR. The Genetics of Hidradenitis Suppurativa. Dermatol Clin. 2016;34(1):23–28.
- Wang B, Yang W, Wen W, et al. Gamma-secretase gene mutations in familial acne inversa. Science. 2010;330(6007):1065.
- Frew JW, Vekic DA, Woods J, Cains GD. A systematic review and critical evaluation of reported pathogenic sequence variants in hidradenitis suppurativa. Br J Dermatol. 2017177(4):987–998.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: results from two case-control studies. J Am Acad Dermatol. 2008;59(4):596–601.
- Sabat R, Chanwangpong A, Schneider-Burrus S, et al. Increased prevalence of metabolic syndrome in patients with acne inversa. PLoS One. 2012;7(2):e31810.
- Kromann CB, Deckers IE, Esmann S, et al. Risk factors, clinical course and long-term prognosis in hidradenitis suppurativa: a cross-sectional study. Br J Dermatol. 2014;171(4):819–824.
- Revuz JE, Jemec GBE. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34(1):1–5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133(6):1506–1511.
- Braun-Falco M, Kovnerystyy O, Lohse P, Ruzicka T. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66(3):409–415.
- Garzorz N, Papanagiotou V, Atenhan A, et al. Pyoderma gangrenosum, acne, psoriasis, arthritis and suppurative hidradenitis (PAPASH)-syndrome: a new entity within the spectrum of autoinflammatory syndromes?. J Eur Acad Dermatol Venereol. 2016;30(1):141–143.
- Vinkel C, Thomsen SF. Autoinflammatory syndromes associated with hidradenitis suppurativa and/or acne. Int J Dermatol. 2017;56(8):811–818.
- Zouboulis CC, Del Marmol V, Mrowietz U, et al. Hidradenitis suppurativa/acne inversa: criteria for diagnosis, severity assessment, classification and disease evaluation. Dermatology. 2015;231(2):184–190.
- Wortsman X. Imaging of Hidradenitis Suppurativa. Dermatol Clin. 2016;34(1):59–68.
- Wortsman X, Moreno C, Soto R, et al. Ultrasound in-depth characterization and staging of hidradenitis suppurativa. Dermatol Surg. 2013;39(12):1835–1842.
- Pascual JC, González I, Corona D, et al. Assessment of subclinical atherosclerosis in hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2017;31(7):1229–1238.
- Tzellos T, Zouboulis CC, Gulliver W, et al. Cardiovascular disease risk factors in patients with hidradenitis suppurativa: a systematic review and meta-analysis of observational studies. Br J Dermatol. 2015;173(5):1142–1155.
- Vinkel C, Thomsen SF. Risk factors for cardiovascular disease in patients with hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2017;31(9):e411–e413.
- Bassas-Vila J, González Lama Y. Hidradenitis suppurativa and perianal Crohn disease: differential diagnosis. Actas Dermosifiliogr. 2016;107 Suppl 2:27–31.
- Downs A. Smoothbeam laser treatment may help improve hidradenitis suppurativa but not Hailey-Hailey disease. J Cosmet Laser Ther. 2004;6(3):163–164.
- van der Zee HH, de Winter K, van der Woude CJ, Prens EP. The prevalence of hidradenitis suppurativa in 1093 patients with inflammatory bowel disease. Br J Dermatol. 2014;171(3):673–675.
- van der Zee HH, van der Woude CJ, Florencia EF, Prens EP. Hidradenitis suppurativa and inflammatory bowel disease: are they associated? Results of a pilot study. Br J Dermatol. 2010;162(1):195–197.
- Hsiao JL, Antaya RJ, Berger T, et al. Hidradenitis suppurativa and concomitant pyoderma gangrenosum: a case series and literature review. Arch Dermatol. 2010;146(11):1265–1270.
- Leuenberger M, Berner J, Di Lucca J, et al. PASS syndrome: an IL-1-driven autoinflammatory disease. Dermatology. 2016;232(2):254–258.
- Miller IM, Ellervik C, Vinding GR, et al. Association of metabolic syndrome and hidradenitis suppurativa. JAMA Dermatol. 2014;150(12):1273–1280.
- Onderdijk AJ, Van Der Zee HH, Esmann S, et al. Depression in patients with hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2013;27(4):473–478.
- Shavit E, Dreiher J, Freud T, et al. Psychiatric comorbidities in 3207 patients with hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2015;29(2):371–376.
- Kurek A, Johanne Peters EM, Sabat R, et al. [Depression is a frequent co-morbidity in patients with acne inversa]. J Dtsch Dermatol Ges. 2013;11(8):743–749, 743–750. Article in German.
- Gulliver W, Zouboulis CC, Prens E, et al. Evidence-based approach to the treatment of hidradenitis suppurativa/acne inversa, based on the European guidelines for hidradenitis suppurativa. Rev Endocr Metab Disord. 2016;17(3):343–351.
- Ingram JR, Woo PN, Chua SL, et al. Interventions for hidradenitis suppurativa: a Cochrane systematic review incorporating GRADE assessment of evidence quality. Br J Dermatol. 2016;174(5):970–978.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22(5):325–328.
- Fischer AH, Haskin A, Okoye GA. Patterns of antimicrobial resistance in lesions of hidradenitis suppurativa. J Am Acad Dermatol. 2017;76(2):309–313.e2.
- Riis PT, Boer J, Prens EP, et al. Intralesional triamcinolone for flares of hidradenitis suppurativa (HS): a case series. J Am Acad Dermatol. 2016;75(6):1151–1155.
- Jemec GB, Wendelboe P. Topical clindamycin versus systemic tetracycline in the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1998;39(6):971–974.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219(2):148–154.
- van der Zee HH, Boer J, Prens EP, Jemec GBE. The effect of combined treatment with oral clindamycin and oral rifampicin in patients with hidradenitis suppurativa. Dermatology. 2009;219(2):143–147.
- Mendonca CO, Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154(5):977–978.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375(5):422–434.
- Grant A, Gonzalez T, Montgomery MO, et al. Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: a randomized, double-blind, placebo-controlled crossover trial. J Am Acad Dermatol. 2010;62(2):205–217.
- Tzanetakou V, Kanni T, Giatrakou S, et al. Safety and efficacy of anakinra in severe hidradenitis suppurativa: a randomized clinical trial. JAMA Dermatol. 2016;152(1):52–59.
- Gulliver WP, Jemec GBE, Baker KA. Experience with ustekinumab for the treatment of moderate to severe Hidradenitis suppurativa. J Eur Acad Dermatol Venereol. 2012;26(7):911–914.
- Blok JL, Li K, Brodmerkel C, et al. Ustekinumab in hidradenitis suppurativa: clinical results and a search for potential biomarkers in serum. Br J Dermatol. 2016;174(4):839–846.
- Matusiak ?, Szcz?ch J, Bieniek A, et al. Increased interleukin (IL)-17 serum levels in patients with hidradenitis suppurativa: implications for treatment with anti-IL-17 agents. J Am Acad Dermatol. 2017;76(4):670–675.
- Mehdizadeh A, Hazen PG, Bechara FG, et al. Recurrence of hidradenitis suppurativa after surgical management: a systematic review and meta-analysis. J Am Acad Dermatol. 2015;73(5 Suppl 1):S70–S77.