J Clin Aesthet Dermatol. 2019;12(12):44–46
 by Mary K. Dick, BS; Michael H. Klug, MD, PharmD; Padma P. Gummadi, MD; Laura K. Klug, PharmD; and Christopher J. Huerter, MD
by Mary K. Dick, BS; Michael H. Klug, MD, PharmD; Padma P. Gummadi, MD; Laura K. Klug, PharmD; and Christopher J. Huerter, MD
Ms. Dick is with the Creighton School of Medicine in Omaha, Nebraska. Drs. M. H. Klug and Gummadi are with the Internal Medicine Department at CHI Health Creighton University Medical Center in Omaha, Nebraska. Dr. L. K. Klug is with the Pharmacy Practice Department at Creighton University School of Pharmacy and Health Professions in Omaha, Nebraska. Dr. Huerter is with the Dermatology Department at CHI Health Creighton University Medical Center in Omaha, Nebraska.
FUNDING: No funding was provided for this study.
DISCLOSURES: The authors have no conflicts of interest relevant to the content of this article.
ABSTRACT: Gardner-Diamond syndrome (GDS) is a psychological and dermatologic syndrome involving painful, ecchymotic, purpuric lesions that typically appear after a period of stress or minor trauma. This syndrome most commonly occurs in young women, though it has also been less commonly reported in men and adolescents. It is an uncommon condition and appropriate diagnosis is important to properly manage symptoms and minimize risks to patients. Here report a case of GDS presenting in a woman with common variable immunodeficiency (CVID), which, to the best of our knowledge, is not a comorbid condition that has previously been reported in correlation with GDS.
KEYWORDS: Autoerythrocyte sensitization, common variable immunodeficiency, Gardner-Diamond syndrome, psychogenic purpura
Gardner-Diamond syndrome (GDS) is a psychological and dermatologic syndrome presenting as painful, ecchymotic, purpuric lesions that typically occur after a period of stress or minor trauma.1 This syndrome has a predisposition among young adult women, but reports concerning men and children have also been published in the literature.2 In 1927, psychiatrist Rudolf Schindler first reported skin hemorrhages suggested to be correlated with hypnosis. One year later, reports describing purpura combined with hysteria and delusions strengthened the link between the psychology and the skin lesions.3 In 1955, GDS was branded by Frank Gardner and Louis Diamond, who identified and discussed four women who developed continuous bruising with local pain followed by erythema and swelling after little or no trauma.1 Gardner and Diamond proposed that the pathophysiology of this condition was related to the autosensitization of patients to their own blood. This finding led to the term autoerythrocyte sensitization syndrome, which is also used to describe GDS. Here, we present a case of GDS appearing in a woman with common variable immunodeficiency (CVID), a cocondition that, to our knowledge, has not been previous described in correlation in the literature.
Case Presentation
A 32-year-old woman with a history of depression, fibromyalgia, and CVID treated with monthly intravenous immunoglobulin (IVIG) presented to the emergency department for pleuritic chest pain, fevers, general fatigue, and edema involving the hands and feet. A chest X-ray demonstrated bronchiectasis. Intravenous methylprednisolone, piperacillin-tazobactam, ipratropium-albuterol, and daily chest physiotherapy were initiated. Bilateral lower-lobe pneumonia was diagnosed three days later. Her scheduled dose of IVIG was delayed due to acute illness.
The patient had previously been taking fluoxetine for depression, but had not been taking it for six months prior to the onset of GDS. One week after her hospital admission, she developed anxiety and clonazepam was started. Three days later, the patient developed areas of scattered ecchymosis on her chest, back, and extremities with allodynia (Figures 1A–1C). Rheumatology was consulted for potential vasculitis. Workup included perinuclear antineutrophil cytoplasmic antibody, antinuclear antibody, angiotensin-converting enzyme, glomerular basement membrane antibodies, prothrombin time, partial thromboplastin time, international normalized ratio, and sedimentation rate. All results came back unremarkable, indicating that small-vessel vasculitis or systemic vasculitis were unlikely.
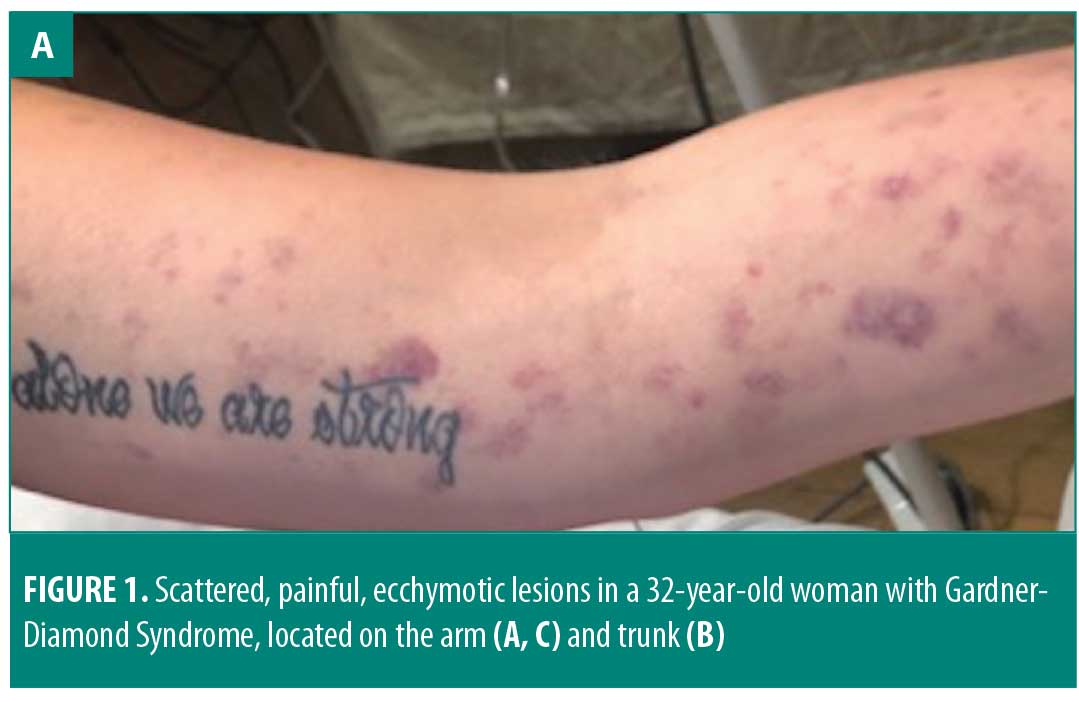
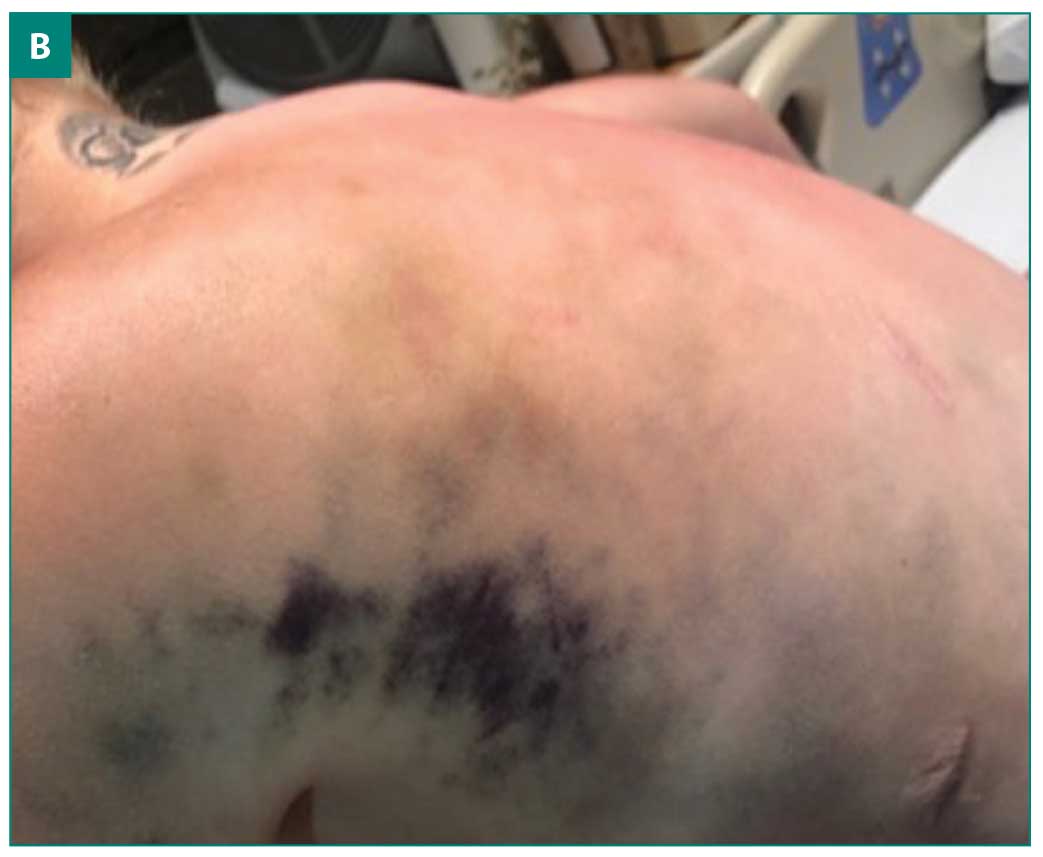
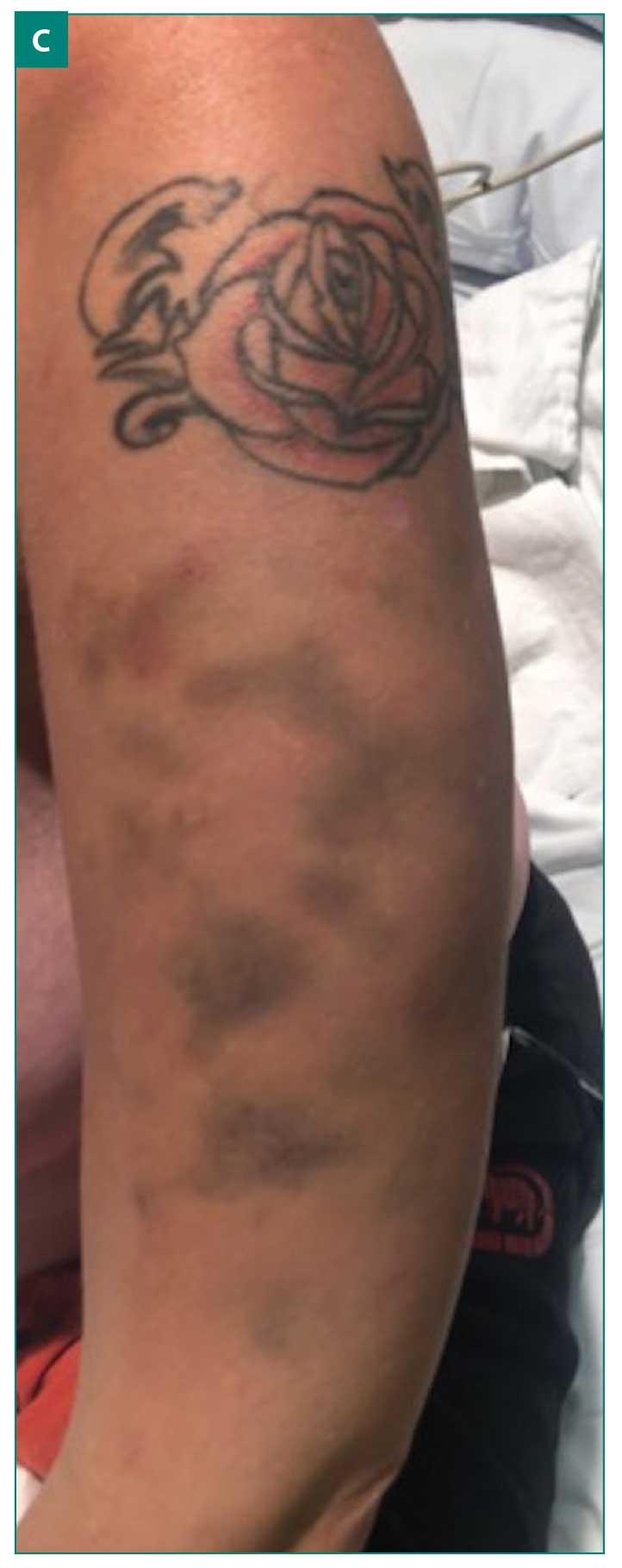 Dermatology was consulted and a diagnosis of GDS was proposed. A punch biopsy showed scattered areas of red blood cell extravasation in the reticular dermis and subcutaneous tissue, with a conspicuous lack of inflammatory infiltrate. These findings are consistent with GDS. Psychiatry was consulted for further anxiety and major depression management.
Dermatology was consulted and a diagnosis of GDS was proposed. A punch biopsy showed scattered areas of red blood cell extravasation in the reticular dermis and subcutaneous tissue, with a conspicuous lack of inflammatory infiltrate. These findings are consistent with GDS. Psychiatry was consulted for further anxiety and major depression management.
Discussion
Pathophysiology. It is typical for GDS to have a relapsing and remitting course with variable intervals between painful occurrences. Lesions can occur anywhere on the body, but most occur on the extremities. Additional associated findings, including myalgias, headaches, and bleeding have been reported.4 To date, many possible pathophysiological mechanisms of GDS have been proposed. Groch et al5 initially found autosensitization directed towards phosphatidylserine present on the erythrocyte cell membrane. However, when phosphatidylserine was later isolated and injected into patients, no reaction was found, leaving room for additional etiologies to be proposed. The role of psychological stress plays a profound part in this condition, with relapses often occurring after an acutely stressful event that alters the hemostatic equilibrium via unclear mechanisms.3 Some proposed mechanisms include increased oxidative damage in patients suffering from depression and increased vascular permeability due to stress induced mast cell degranulation.6 A relationship between estrogen and this condition has also been suggested, given that women are more commonly affected.4
Our patient’s past medical history of CVID is a unique comorbidity in this context. CVID is a primary immunodeficiency involving an antibody production defect. This disorder increases the patient’s risk of infection, cancer, and autoimmune conditions. Up to 20 percent of patients with CVID will be affected by an autoimmune disorder, with the most common being immune thrombocytopenic purpura.7,8 The proliferation of autoimmune cells is thought to be caused by a combination of immune dysregulation coupled with repeated immune system stimulation secondary to frequent infections. These fluctuations in immune system regulation could be a factor in the development of GDS.8
Diagnosis. GDS is a diagnosis of exclusion after ruling out bleeding disorders, including Von Willebrand disease, hemophilia, idiopathic thrombocytopenic purpura, Henoch-Schönlein purpura, and systemic or cutaneous vasculitis.3 No specific laboratory tests can confirm a GDS diagnosis.9 One controversial test involves injecting the patient with their own plasma, using normal saline as the control. Bruising limited to the site of plasma injection is considered a positive result supporting a GDS diagnosis. However, this test has limited specificity and sensitivity, and positive responses to the control substance or negative responses to plasma do not completely rule out GDS. Also, performing this test can complicate the diagnosis and cause confusion for the patient, so its use is limited and not generally recommended.10
Ultimately, the diagnosis is made following a detailed medical and psychiatric history, physical examination, and exclusion of other possible causes. Skin biopsy to rule out vasculitis is of benefit. The accurate diagnosis of GDS is particularly important to limit unnecessary procedures in affected patients. While operations on patients with GDS have been successfully completed without major bleeding complications, surgery should be limited or even contraindicated, per some literature, in patients with a GDS diagnosis.9
Treatment. Currently, GDS lacks definitive treatment options. Various antihistamines, corticosteroids, hormonal contraceptives, antibiotics, and vitamin C have been tested with minimal efficacy and with some patients reporting positive results from placebos.4 A multidisciplinary approach focused on improving underlying psychological conditions plays an integral role in effective treatment. Most patients benefit from a combination of cognitive behavioral therapy and antidepressant treatment.3
Conclusion
GDS is an uncommon condition involving spontaneous and unexplained painful ecchymosis and purpura, typically with a psychological association. A timely and accurate diagnosis is necessary to avoid excessive and unnecessary treatments. Since the diagnosis is done by exclusion and no existing laboratory tests significantly aid in the diagnosis, a thorough psychiatric history and evaluation should be completed, with other probable causes ruled out before confirming GDS.
References
- Gardner FH, Diamond LK. Autoerythrocyte sensitization A form of purpura producing painful bruising following autosensitization to red blood cells in certain women. Blood. 1995;10(7): 675–690.
- Ivanov O, Lvov A, Michenko A, et al. Autoerythrocyte sensitization syndrome (Gardner-Diamond syndrome): review of the literature. J Eur Acad Dermatol Venereol. 2009;23(5):4999–4504.
- Jafferany MMD, Bhattacharya GMD. Psychogenic purpura (Gardner-Diamond syndrome). Prim Care Companion CNS Disord. 2015;17(1).
- Karakas Z, Karaman S, Avci B, et al. A disease difficult to diagnose: Gardner-Diamond syndrome accompanied by platelet dysfunction. Turk Pediatri Ars. 2014;49(3):250–253.
- Groch G, Finch S, Rogoway W, Fischer D. Studies in the pathogenesis of autoerythrocyte sensitization syndrome. Blood. 1966;28(1):19–33.
- Cansu D, Kasifoglu T, Pasaoglu O, Korkmaz C. Autoerythrocyte sensitization syndrome (Gardner-Diamond syndrome) associated with cutaneous vasculitis. Joint Bone Spine. 2008;75(6):721–724.
- Tainwala RR, Phiske M, Raghuwanshi A, et al. Perplexing purpura in two females: rare case of autoerythrocyte sensitization syndrome. Indian Dermatol Online J. 2013;4(4):305–308.
- Meeder R, Bannister S. Gardner-Diamond syndrome: Difficulties in the management of patients with unexplained medical symptoms. Paediatr Child Health. 2006;11(7):416–419.
- Jesus AA, Jacob CMA, Silva CA, et al. Common variable immunodeficiency associated with hepatosplenic T-cell lymphoma mimicking juvenile systemic lupus erythematosus. Clin Dev Immunol. 2011;2011:428703.
- Patuzzo G, Barbieri A, Tinazzi E, et al. Autoimmunity and infection in common variable immunodeficiency (CVID). Autoimmun Rev. 2016;15(9):877–882.