Nicholas Schmidt, PhD; Eugene H. Gans, PhD
Drs. Schmidt and Gans are from Medicis Pharmaceutical Corporation, Scottsdale, Arizona
Disclosure: The preparation of this manuscript was funded by Medicis Pharmaceutical Corporation, Scottsdale, Arizona. Drs. Schmidt and Gans are consultants for Medicis Pharmaceutical Corporation and have no financial interest.
Abstract
Tretinoin has been primarily used for the early stages of acne because of its proven comedolytic end benefits. This article reviews and updates the collective body of evidence of tretinoin in the treatment of acne, which suggests that this drug also possesses a broad range of acne-related immunomodulating properties that are capable of disrupting and hindering the various stages of the inflammatory cascade and the production of proinflammatory factors associated with it.
(J Clin Aesthet Dermatol. 2011;4(11):22–29.)
Historically, tretinoin (all-trans retinoic acid) has been used as a comedolytic agent to treat mild-to-moderate acne. Used as a stand-alone treatment or in combination with antibacterials, its ability to stimulate the growth of new cells, unclog pores, and promote the normal flow of sebum is well-proven. However, over the years there has been a growing awareness within the professional community that tretinoin also possesses a broad range of modulating properties, which could play a beneficial role in the disruption of the immunoinflammatory cascade of acne vulgaris and the proinflammatory factors associated with it. This review article is a compilation of such data and is intended to provide the researcher and practitioner with a greater appreciation of the expansive role that tretinoin plays in the treatment of inflammatory acne.
Inflammatory Acne
Acne vulgaris is a chronic inflammatory disease of a pilosebaceous unit that is able to respond to the threat of infection as an immunocompetent organ.[1] It is generally thought that hypercornification, plugging of the follicular opening by sloughed keratinocytes and other debris, comedone formation, excess sebum production, abnormal proliferation of Propionibacterium acnes, and leukocyte infiltration combine to set the stage for a localized inflammatory response.[2–6]
The sebaceous follicle is sensitive to any changes that signal the beginning of an inflammatory episode. Although not fully understood, the first sign of subclinical acne is thought to be the microcomedo “lesion” within which abnormal amounts of keratinized cells are produced. Keratinocytes are part of the skin’s first line of defense and, in conjunction with sebocytes, produce proinflammatory chemokines and cytokines that alert the host to the presence of danger and attract lymphocytes and T-helper cells to the microbially infected area.[7] This initial event appears to be the onset of an intracellular communication/ signaling process that provides various tissues and organs with infectious information that determines what protective immunological steps to take next. If it is a typical acne inflammatory event, an innate (nonspecific) and acquired (antibody-mediated) immune response will occur.
The ensuing overgrowth of P. acnes within the plugged follicle results in the discharge of extracellular foreign materials by this organism. These proinflammatory triggers include lipase, which releases cytotoxic free fatty acids from sebum, as well as protease, hyaluronidase, and neuraminidase—all of which can damage the follicular wall, promote leakage into the surrounding dermis, and trigger inflammation.[8–14] P. acnes is also capable of producing other undesirable proinflammatory agents, such as highly immunogenic heat shock proteins, porphyrin that reacts with molecular oxygen to produce toxic oxygen species and free radicals, and highly comedogenic squalene peroxides.[5,15–17]
During the early stages of preinflammatory and inflammatory acne, P. acnes releases potent low molecular weight chemotactive factors and chemotactic lipase that find their way into the dermis and surround the affected follicle.[18–23] The latter are capable of attracting various white blood cells to the site of infection, such as neutrophils, lymphocytes, and monocytes.[3,13,19,24–27]
P. acnes also stimulates the production of small polypeptide, proinflammatory cytokines during the onset of infection. They include interleukins (IL) IL-1a, IL-1b, IL-6, IL-8, and IL-12, granulocyte-macrophage colony-stimulating factor (GM-CSF), tumor necrosis factor alpha (TNF-a), and interferon-gamma (IFN-g). These biologically active messenger molecules originate from white blood cells, keratinocytes, and sebocytes in and around the damaged follicle, as the host cross-talks with skin defense and immune cells to fight and ward off the infection.[1,17,28–39]
Along with the above inflammatory challenges, other innate and acquired immune responses are called into play by the host, which add to the cascade of proinflammatory events within the distended, leaky follicle. Immunoactive T lymphocytes (T-cells), CD+4 cells, and CD14+ cells infiltrate the inflamed lesion.[1,36,40–43] Activated monocytes transform into macrophages and begin to surround and engulf foreign materials associated with the infectious threat. Intracellulary, phagocytosing neutrophils/macrophages release degradative lysosomal enzymes and toxic chemicals, such as reactive oxygen species, all of which help to break down and remove P. acnes and its injurious extracellular products.[44–47] When produced chronically and excessively, these same materials may also rupture the follicle, find their way into the surrounding dermis, and be inflammatory to the host.[5,46,48–51]
Other proinflammatory factors that are thought to be implicated with pathogenic acne are leukotriene B4 (LTB4) and compliment-activated C5-derived neutrophil chemotactic factors, all of which can recruit and activate neutrophils, monocytes, and eosinophils resulting in more inflammation.[40,52–55] A host of other inflammatory agents and events likely add to the proinflammatory fray: toll-like receptors (TLRs), transcription factor activator protein 1 (AP-1), vascular cell adhesion, macrophage degranulation, growth factors, expression of transglutaminase and lipoxygenase, cytokine modulation of metalloproteinase activity in the dermis, prostaglandin synthesis, and the possible release of IL-5.[30,33,36,43,56–62]
At some point, the inflamed lesion may burst and empty its bacterial contents, cytokines, and chemotactic factors into the dermis and epidermis, which is considered to be the most distressing event in the course of inflammatory acne. This continues to exacerbate the cascade of inflammation within and outside the immediate environment of the ruptured follicle. The net clinical result that typically occurs is moderate-to-severe acne characterized by papules, pustules, and, in more inflamed cases, nodules.
Tretinoin’s Comedolytic Activities and Potential Anti-inflammatory End Benefits
The comedolytic-related activity of tretinoin has been known for many years. Tretinoin a) normalizes the exfoliation of the follicular epithelium within the pilosebaceous unit, b) inhibits transglutaminase activity that results in cellular adhesion, c) prevents follicular plugging, d) leads to drainage and expulsion of excess sebum and P. acnes, and e) helps create a more aerobic environment that is less conducive to P. acnes.[2,56,63–67] Although they are often thought of as “indirect” effects, the aforementioned therapeutic end benefits can be considered anti-inflammatory, since all of them help to diminish the potential severity and outcome of an inflamed lesion.
Phagocytosis and the Immunomodulating Properties of Tretinoin
Phagocytosis plays a key role in the defense against bacterial infections including acne. Although inflammatory by nature, it is a necessary part of the host’s mechanism of protection. When taken as a whole, some authors contend that homeostatic phagocytosis might be considered an anti-inflammatory response since removal of harmful bacteria, their extracellular byproducts, and other proinflammatory debris is beneficial to the host.[56,68,69] Others suggest that during bouts of excessive inflammation, phagocytes can break down and release inflammatory lysozymes and various reactive oxygen species, which can exacerbate inflammation.[44–47] A recent article points out that macrophages can initially add to the inflammatory process, but, when activated to another form, may actually aid in the healing process. This would explain how untreated inflamed lesions resolve favorably and return to normalcy on their own.[70] Overall, the net effect of macrophages in the role of infection may very well be considered a positive one and is treated as such in this article.
Five different inflammatory events associated with acne and phagocytosis are phagocytic function, respiratory burst, inducible nitrous oxide synthase (iNOS) activity, degranulation, and proteolytic enzyme activity. Available data suggest that tretinoin beneficially modulates all of them.
Phagocytic function. Retinoic acid has been shown to modulate in-vitro macrophage function. When immuno-globulin G-sensitized bovine erythrocytes were exposed to a mouse macrophage cell line, tretinoin increased phagocytosis more than twofold at low concentrations (5 x 10-11M) and higher when compared to controls. The authors state that they have also observed similar results in vivo.[71]
Respiratory burst. Several studies have been conducted which showed that tretinoin is able to suppress respiratory burst and its release of cytotoxic oxygen species, such as superoxide anion and hydrogen peroxide, which could result in less tissue irritation.[59,60,72,73] The first study employed human polymorphonucleocytes and compounds known to activate a respiratory burst response. At nontoxic concentrations, tretinoin produced a dose-dependent (1 to 100µM) inhibition of superoxide anion that reached suppression levels up to 90 percent. The investigators suggest that tretinoin’s dampening effect could explain, in part, its anti-inflammatory properties.[59] A second study showed that tretinoin produced a medium-to-potent inhibition of oxidative burst in rabbit-derived poly-morphonucleocytes.60 And two in-vitro studies showed that tretinoin significantly inhibited the release of oxygen free radicals and the generation of superoxide anions.[72,73]
Nitrous oxide synthase. Proinflammatory cytokines are able to upregulate the cellular expression of nitric acid synthase (iNOS enzyme).[73] Using various forms of this enzyme, keratinocytes and macrophages can produce nitric oxide, which is an important mediator for inflammatory reactions and may even be responsible for triggering the inflammatory cascade.[15]
Tretinoin is known to suppress the release of cytotoxic nitric oxide. Using activated human keratinocytes, investigators showed that tretinoin was able to downregulate the production of nitrites (reflecting nitric oxide pathway activation) up to 70 percent in a dose-dependent manner. In the same test model, the investigators also showed that this same retinoid inhibited the induction of iNOS by 60 percent and suppressed the expression of iNOS messenger RNA (mRNA) by 50 to 70 percent.[74] In a similar study several years later, investigators determined the effect of tretinoin on nitric acid release, induction of iNOS, and expression of iNOS mRNA using human keratinocytes and immuno-globulin E as the induction agent. The results showed a time- and dose-dependent inhibition effect, with the highest levels of suppression being 60 percent for iNOS activity and, after 24 hours of treatment, virtually eliminated the expression of iNOS mRNA.[75]
Degranulation. Degranulation occurs when intracellular granules from phagocytic cells, such as immunocytes and neutrophils, release degradative lysozymes that can digest infectious organisms including P. acnes, which can be irritants if released. Two in-vitro studies showed that tretinoin inhibits degranulation in phagocytic cells resulting in a diminished release of cytotoxic, tissue-irritating materials.[50,59]
Proteolytic enzymes. During phagocytosis, macro-phages engulf and degrade harmful micro-organisms using proteolytic enzymes. The latter can find their way into the surrounding microenvironment of the acne lesion and cause inflammation. The inhibitory effect of tretinoin on proteolytic enzyme release in human neutrophils showed that increasing concentrations of this drug (5 to 20µg) resulted in correspondingly smaller levels of the degradative enzymes when compared to untreated controls.[73]
Tretinoin’s Cytokine Immunomodulating Effects
Cytokines are transient immunomodulating substances secreted by specific cells of the immune system. Capable of carrying messages between local cells via signaling pathways, they regulate the duration and amplitude of the immune response. Normally found in unstressed sebaceous glands, their levels increase dramatically when an infectious P. acnes challenge occurs and they become key players in the ensuing inflammatory cascade.[76–78]
IL-5 is a cytokine that appears to play a beneficial role in the immunomodulatory process. It has been suggested that upregulation of IL-5 can activate specific T-helper (Th) lymphocytes, such as Th2 cells. These cells, based on their cytokine profiles, may actually aid in the healing of acne lesions. Tretinoin has been shown to activate IL-5 in human peripheral blood mononuclear cells.[62]
IL-6 is a proinflammatory mediator that may play an important role during the early stages of acne. Macrophages, T-cells, and keratinocytes can produce IL-6 during an infectious challenge.[35,79] Using human epidermoid carcinoma A431 cells, tretinoin exhibited a “very potent” inhibition of the phorbol 12-myristate 13-acetate-stimulated release of proinflammatory IL-6 when compared to control cells.[62] A second study confirmed these observations when CD14+ monocytes, the predominant form of TLR2+ cells found in the study of acne lesions, were exposed to heat-killed P. acnes. Tretinoin demonstrated a 74-percent inhibition of the release of IL-6 in the monocytes.[43]
IL-12 is produced by macrophages in the presence of antigenic challenge. During an inflammatory episode, it stimulates the growth and function of various immunocytes (especially T cells), the production of IFN-g and TNF-a, and plays an important induction role in the Th1 immune response. It has been shown that mouse macrophages can produce IL-12 when stimulated with a known lipopolysaccharide (LPS) antigen or the heat-killed pathogenic organism, Listeria monocytogenes. When these same macrophages were pretreated with varying amounts of tretinoin, the induction of proinflammatory IL-12 was significantly reduced in a dose-dependent manner, decreasing from approximately 840pg/mL to about 420pg/mL when compared to untreated controls. Administration of tretinoin also caused mouse macrophages to increase production of IL-4 by antigen-primed CD4+ T cells and helped these cells differentiate towards a Th2 phenotype, known to promote healing.[80]
IL-12 is composed of p35 and p40 protein subunits. IL-12-p40 proinflammatory mediators can be released in varying amounts independent of IL-12 and can induce the Th1 helper cell inflammatory response. A recent in-vitro study measured the inhibitory effects of tretinoin on the secretion of IL-12-p40 in human monocytes using heat-killed P. acnes to activate these white blood cells. Results showed that tretinoin was able to downregulate the induction of IL-12-p40 by 91 percent.[43]
TNF-a, produced in part by human keratinocytes, is a multifunctional cytokine that can induce a broad range of secondary proinflammatory effects in response to microbial infections. It can activate cell adhesion molecules; upregulates prostaglandins, collagenase, various inflammatory cells, and human acute monocytic leukemia cell line (ThP-1) cells; and can induce the release of other cytokines, such as IL-1, IL-6, IL-8, and granulocyte-macrophage colony-stimulating factor.[28,31,39]
Several studies have shown that tretinoin is a potent inhibitor of TNF-a production.[43,74,75] The first study evaluated the effects of tretinoin on the release of TNF-a in CD14+ human monocytes, wherein heat-killed P. acnes served as the antigenic stimulant. Results showed that tretinoin was able to downregulate the induction of TNF-a by 70 percent.43 In a second study, investigators evaluated the effects of tretinoin on the production of TNF-a secretion by human keratinocytes activated with lipopolysaccharides and IFN-g. In a dose-dependent fashion, tretinoin inhibited the release of TNF-a up to 60 percent.[74] Another similar investigation measured the inhibitory effect of tretinoin on TNF-a release by human keratinocytes. Immunoglobulin E was used as the induction agent to stimulate the release of TNF-a. Results showed a time- and dose-dependent inhibition effect, with the greatest level of TNF-a inhibition being approximately 70 percent.[75]
IFN-g, another potent cytokine, primes macrophages making them cytotoxic, increases antigen presentation of macrophages, induces the production of E-selectin on endothelial cells, and the production of intercellular adhesion molecule-1 and human leukocyte antigen complex (HLA-DR) on keratinocytes. All of these actions play an important role in the proinflammatory attraction of leukocytes to the site of infection.[81–83] The in-vitro inhibitory effect of tretinoin on IFN-g expression was evaluated in normal human peripheral blood mononuclear leukocytes activated in the presence of Staphylococcus enterotoxin B. Tretinoin “very potently” inhibited the release of the proinflammatory cytokine, IFN-g.[62] A second study evaluated the effect of tretinoin on the induction of IFN-g by mouse macrophages, which were stimulated with LPS antigen or a heat-killed pathogenic organism, Listeria monocytogenes. Macrophages pretreated with varying concentrations of tretinoin exhibited approximately one-half the induction of proinflammatory IFN-g compared to control values.[80]
Tretinoin’s Effect on Other Important Proinflammatory Events
Other important proinflammatory events take place during the inflammatory cascade, all of which may be beneficially modulated by tretinoin. LTB[4] is a proinflammatory agent that is able to recruit neutrophils and other immunocytes to the site of infection and plays a role in stimulating the production of various proinflammatory cytokines and other bioactive mediators. Retinoids have been shown to be potent inhibitors of rat leukocyte LTB4 production in vitro. The following three methods were used to measure the effect of tretinoin on LTB[4 release in rat leukocytes: 1) polymorphonucleocyte aggregation, 2) polymorphonucleocyte chemokinesis, and 3) physico-chemical measurements as reverse phase-high pressure chromatography. All three methods showed that tretinoin was a potent inhibitor of LTB4 production.[84]
T helper cells, such as CD4+ T-helper lymphocyte cells are among the earliest to infiltrate the lesion during the early stages of acne inflammation. The Th1 subset of these cells are known to secrete proinflammatory cytokines, such as IFN-g, TNF-a, and others.[62,80] It has also been suggested that the subset of Th2 helper cells might provide healing influences in acne lesions.[62,85] In a study that evaluated the effect of tretinoin on the expression of IL-5 and TNF-a, it was found that the production of IL-5 was significantly increased while that of TNF-a was inhibited. The investigators suggested that this dual effect could in turn increase the influence of Th2 cells, reduce the response of Th1 cells, and thereby promote healing.[62]
Immunocyte toll-like receptors recognize and identify molecular patterns that occur on invasive bacteria after which they upregulate the induction of IL-1 and helper T cells.[35,56] Several studies have demonstrated that tretinoin can suppress the induction of toll-like receptors.[43,86] One study measured the effects of tretinoin on the expression of toll-like receptor-2 (TLR-2) receptor sites in human monocytes. Results showed that tretinoin was able to downregulate the activation of TRL-2 by approximately 65 percent as measured by the decrease in mRNA that controls this expression. The activation of the coreceptor of TRL-2, known as CD14, was also inhibited by 34 percent. However, tretinoin had no effect on the expression of TRL-1 and TRL-4 receptor sites.[43] A second in-vitro study showed tretinoin could suppress the expression of TRL-2 in human monocytes. The investigators initially determined that TRL-2 receptor site was expressed by these immune cells and that the TRL-4 site was not. The authors state, in part, that “all-trans retinoic acid was also active” in suppressing the expression of TRL-2 in human monocytes.[86]
Transcription factor AP-1 is used by modulators of inflammation to control the orchestration and expression of several genes involved with the inflammatory process. Two of these genes encode for vascular endothelial growth factor and matrix metalloproteins. An in-vitro study measured, in part, the effects of tretinoin on the expression of transcription factor AP-1. Results showed that the extent of inhibition by tretinoin approached 60 percent.[57]
Cell adhesion protein molecules bind leukocytes to endothelial cells at the site of infection during an initial infectious microbial challenge. It is one of the first defensive immune steps taken by the host. Three different cell adhesion protein molecules are involved in this process, one of which is vascular cell adhesion molecule-1 (VCAM-1). The regulated expression of cell adhesion molecules plays a central role in the initiation of acute and chronic inflammatory disorders. Investigators evaluated, in part, the effect of tretinoin on TNF-a–induced expression of VCAM-1 by cultured dermal microvascular endothelial cells. The authors concluded that “The specific inhibition of cytokine-mediated VCAM-1 gene expression in vitro may provide a potential basis by which retinoids exert their biological effects at sites of inflammation in vivo.”[58]
Prostaglandins can have both proinflammatory and anti-inflammatory properties. Prostaglandin E2 (PGE-2) is released by blood vessel walls in response to infection or inflammation and is typically associated with the inflammatory response. Investigators studied the in-vitro production of inflammation-related PGE-2 in cultured cortical astrocytes, which are immune-competent cells found in the central nervous system. LPS was used as an antigen to activate the cortical astrocytes and the latter’s array of PGE-2 synthesizing enzymes. Under these conditions, the production of PGE-2 was dramatically increased. However, when repeated in the presence of retinoic acid, the production of proinflammatory PGE-2 was inhibited by about 60 percent.[87]
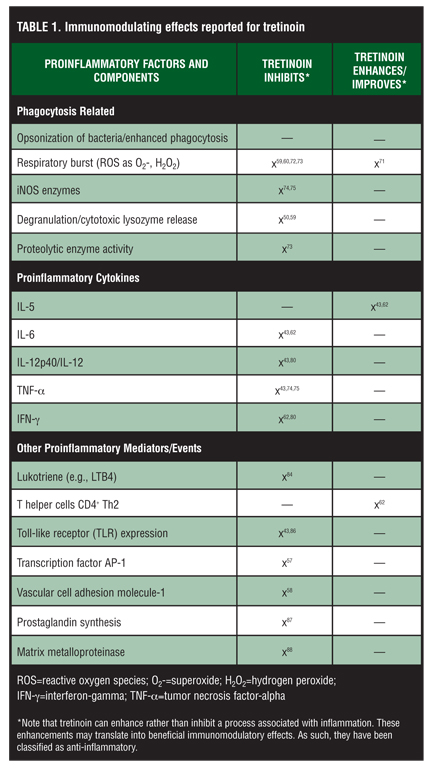
Matrix metalloproteinases (MMPs) break down the supportive structure of the extracellular matrix of tissues. When expressed excessively, they can be inflammatory. It has been shown that P. acnes induces the production of several MMPs, MMP-9 and MMP-1, in primary human monocytes. In vivo, it has been demonstrated that dermal and perifollicular cells can infiltrate acne lesions and express potentially inflam-matory MMP-1 and MMP-9 proteases. In this same study, investigators examined, in part, the in-vitro role that tretinoin might play in the regulation of MMPs. Monocytes exposed to this retinoid significantly decreased the production of MMP-9. When a mixture of mono-cytes, tretinoin, and P. acnes was evaluated, tretinoin inhibited the expression of MMP-9 and MMP-1, while simultaneously increasing the level of TMP metalloproteinase inhibitor 1 (TMP1). The authors noted that, “These data indicate that P. acnes-induced MMPs and TMPs may be involved in acne pathogenesis and that retinoic acid modulates MMP and TMP expression, shifting from a matrix-degradative phenotype to a matrix-preserving pheno-type.”[88] Table 1 summarizes the immuno-modulating observations described for tretinoin.
{kind=link}
Conclusion
The topical use of tretinoin as an anti-acne agent began almost a half century ago. Since that time it has been successfully used to treat comedonal and inflammatory acne. Over the intervening years, the beneficial effects of tretinoin have grown from an understanding of its potent comedolytic-related properties to an evolving appreciation of its anti-inflammatory actions.
This extensive review of tretinoin’s immunomodulating properties suggests that it possesses five important comedolytic-related properties that can be considered anti-inflammatory in nature, as well as 17 other potential activities that may beneficially temper the inflammatory cascade of acne and the production of inflammatory vectors associated with it.
In more recent times, tretinoin has been combined with antibacterials, such as erythromycin, benzoyl peroxide, and clindamycin, to form topical combination treatments for acne. In a recent review of clindamycin’s possible anti-inflammatory properties, as many as 14 possible activities were found that could have immunomodulating effects, any combination of which may beneficially influence the inflammatory response in acne vulgaris.[89] The topical use of clindamycin and tretinoin as a combination treatment modality that includes antibacterial, comedolytic, and anti-inflammatory properties has proven to be a very effective therapy for treating the various stages of acne.[90–96] It is now becoming increasingly clear that there may be good reasons for these observations.
References
1. Koreck A, Pivarcsi, Dobozy A, et al. The role of innate immunity in the pathogenesis of acne. Dermatology. 2003;206:96–105.
2. Berson DS, Shalita AR. The treatment of acne: the role of combination therapies. J Am Acad Dermatol. 1995;32(5 pt 3):S31–S41.
3. Gollnick HP, Zouboulis CC, Akamatsu H, et al. Pathogenesis and pathogenesis related treatment of acne. J Dermatol. 1991;18:489–499.
4. Cunliffe WJ. The sebaceous gland and acne—40 years on. Dermatology. 1998;196:9–15.
5. Toyoda M, Morohashi M. Pathogenesis of acne. Med Electron Micros. 2001;34:29–40.
6. Zouboulis CC, Piquero-Martin J. Update and future of systemic acne treatment. Dermatology. 2003;206:37–53.
7. Jappe U. Pathological mechanisms of acne with special emphasis on Proprionibacterium acnes and related therapy. Acta Derm Venereol. 2003;83:241–248.
8. Hoeffler U. Enzymatic and hemolytic properties of Proprionibacterium acnes and related bacteria. J Clin Microbiol. 1977;6:555.
9. Van Vlem B, Vanholder R, De Paepe P, et al. Immunomodulating effects of antibiotics: literature review. Infection. 1996;24:275–291.
10. Ingham E, Holland KT, Gowland G, et al. Purification and partial characterization of an acid phosphatase (EC 3.1.3.2) produced by Proprionibacterium acnes. J Gen Microbiol. 1980;118:59.
11. Puhvel S, Reisner RM. The production of hyaluronidase (hyaluronate lyase) by Cornybacterium acnes. J Invest. Dermatol. 1972;58:66.
12. Holland KT. Nutrition of resident micro-organisms. In: Noble WC, ed. The Skin Microflora and Microbial Skin Disease. Cambridge, England: Cambridge University Press; 1992:33–72.
13. Burkhart CG, Burkhart CN, Lehmann PF. Acne: a review of immunologic and microbiologic factors. Postgrad Med J. 1999;75:328–331.
14. Thomsen RJ, Stranieri A, Knutson D. Topical clindamycin treatment of acne. Arch Dermatol. 1980;116:1031–1034.
15. Holland KT, Aldana O, Bojar RA, et al. Proprionibacterium acnes and acne. Dermatology. 1998;196:67–68.
16. Farrar MD, Ingham E, Holland ET. Heat shock proteins and inflammatory acne vulgaris: molecular cloning, over-expression and purification of a Proprionibacterium acnes GroEL and DnaK homologue. FEMS Microbiol Lett. 2000;191:183–186.
17. Graham GM, Farrar MD, Cruse-Sawyer JE, et al. Proinflammatory cytokine production by human keratinocytes stimulated with Proprionibacterium acnes and P. acnes GroEL. Br J Dermatol. 2004;150:421–428.
18. Puhvel SM, Sakamoto M. The chemoattractant properties of comedonal components. J Invest Dermatol. 1978;71:324–329.
19. Puhvel SM, Sakamoto M. Cytotaxin production by comedonal bacteria. J Invest Dermatol. 1980;74:36–40.
20. Lee WL, Shalita AR, Sunthralingam K, et al. Neutrophil chemotaxis to Proprionibacterium acnes lipase and its inhibition. Infect Immun. 1982;35:71–78.
21. Webster GF, Leyden JJ. Characterization of serum-independent polymorphonuclear leukocyte chemotactic factors produced by Proprionibacterium acnes. Inflammation. 1980;4;261–271.
22. Webster GF. Inflammation in acne vulgaris. J Am Acad Dermatol. 1995;33(2 Part 1):247–253.
23. Tan H. Topical antibacterial treatments for acne vulgaris. Am J Clin Dermatol. 2004;5(2);79–84.
24. Webster GF, Leyden JJ, McGinley KJ, et al. Suppression of polymorphonuclear leukocyte chemotactic factor production in Proprionibacterium acnes by subliminal inhibitory concentrations of tetracycline, ampicillin, minocycline and erythromycin. Antimicrob Agents Chemother. 1982;21:770–772.
25. Brown SK, Shalita AR. Acne vulgaris. Lancet. 1998;351: 1871–1976.
26. Zouboulis CC. Acne: current aspects on pathology and treatment. Dermatol Experiences. 1999;1:6–37.
27. Piquero-Martin J. Acne Manejo Racional. 3rd ed. Caracas, Venezuela: Corpografica; 2000.
28. Vowels BR, Yang S, Leyden JJ. Induction of proinflammatory cytokines by a soluble factor of Proprionibacterium acnes: implications for chronic inflammatory acne. Infect Immun. 1995;63:3158.
29. Schaller M, Lowenstein M, Borelli C, et al. Induction of chemoattractive proinflammatory cytokine response after stimulation of keratinocytes with Proprionibacterium acnes and coproporphyrin III. Br J Dermatol. 2005;153: 66–71.
30. Luger TA, Schwarz T. Evidence for an epidermal cytokine network. J Invest Dermatol. 1990;95:1005–1011.
31. Kock A, Schwarz T, Kirnbauer R, et al. Human keratinocytes are a source for tumor necrosis factor a: evidence for synthesis and release upon stimulation with endotoxin or ultraviolet light. J Exp Med. 1990;172:1609–1614.
32. Kupper TS. The activated keratinocyte: a model for inducible cytokine production by non-bone marrow-derived cells in cutaneous inflammatory and immune responses. J Invest Dermatol. 1990;94:146S–150S.
33. Chen Q, Koga T, Uchi H, et al. Proprionibacterium acnes-induced IL-8 production may be mediated by NF-kappa‚ activation in human monocytes. J Dermatol Sci. 2002:29:97–103.
34. Basal E, Jain A, Kaushal GP. Antibody response to crude cell lysate of Proprionibacterium acnes and induction of pro-inflammatory cytokines in patients with acne and normal healthy subjects. J Microbiol. 2004;42(2):117–125.
35. Kim J, Ochoa M, Krutzik S, et al. Activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. J Immunol. 2002;169(3):1535–1541.
36. Miller CC, Hale P, Pretland AP. Ultraviolet light injury increases prostaglandin synthesis through a tyrosine kinase-dependent pathway. J Biol Chem. 1994;269:3529–3533.
37. Ingham E, Eady EA, Goodwin CE, et al. Pro-inflammatory levels of interleukin 1a-like bioactivity are present in the majority of open comedones in acne vulgaris. J Invest Dermatol. 1992;98:895–501.
38. Ansel J, Perry P, Brown J, et al. Cytokine modulation of keratinocyte cytokines. J Invest Dermatol. 1990;94(6 Suppl):101S–107S.
39. Kuwahara K, Kitazawa T, Kitagaki H, et al. Nadifloxacin, an antiacne quinolone antimicrobial, inhibits the production of proinflammatory cytokines by human peripheral mononuclear cells and normal human keratinocytes. J Dermatol Sci. 2005;38:47–55.
40. Jappe U, Ingham E, Henwood J, et al. Propionibacterium acnes and inflammation in acne; P. acnes has T-Cell mitogenic activity. Br J Dermatol. 2002;146:202–209.
41. Jeremy HT, Holland DB, Roberts SG, et al. Inflammatory events are involved in acne lesion initiation. J Invest Dermatol. 2003;121(1):20–27.
42. Norris JFB, Cunliffe WJ. A histological and immunological study of early acne lesions. Br J Dermatol. 1988;118: 651–659.
43. Liu PT, Krutzok SR, Kim J, et al. Cutting edge: all-trans-retinoic acid down-regulates TRL2 expression and function. J Immunol. 2005;174:2467–2470.
44. Webster GF, Leyden JJ, Tsai CC, et al. Polymorphonuclear leukocyte lysosomal release in response to Propionibacterium acnes in vitro and its enhancement by sera from patients with inflammatory acne. J Invest Dermatol. 1980;74:398–401.
45. Webster GF, Kligman AM. A method for the assay of inflammatory acne of inflammatory mediators in follicular casts. J Invest Dermatol. 1979;73:266–268.
46. Miyachi Y, Yoshioka A, Imamura S, et al. Effect of antibiotics on the generation of reactive oxygen species. J Invest Dermatol. 1986; 86:449–453.
47. Akamatsu H, Komura J, Miyachi Y, et al. Suppressive effects of linoleic acid on neutrophil oxygen metabolism and phagocytosis. J Invest Dermatol. 1990;95:271–274.
48. Webster GF, McGinley KJ, Leyden JJ. Inhibition of lipase production in Propionibacterium acnes by subminimal inhibitory concentrations of tetracycline and erythromycin. Br J Dermatol. 1981;104:453–457.
49. Akamatsu H, Kurokawa I, Nishijima S, et al. Inhibition of neutrophil chemotactic factor production in comedonal bacteria by subminimal inhibitory concentrations of erythromycin. Dermatology. 1992;185:41–43.
50. Camisa C, Eisenstat B, Ragaz A. The effects of retinoids on neutrophil function in vitro. J Am Acad Dermatol. 1982;6(4):620–629.
51. Dreno B. Modulation of superoxide dismutase and glutathione peroxide enzymes by minocycline: in vitro and in vivo study. Presented at the American Academy of Dermatology, 47th annual meeting; Washington, DC: December 4–9, 1993.
52. Shalita AR, Wei-Li L. Inflammatory acne. Dermatol Clin. 1983;1:361–364.
53. Massey A, Mowbray JF, Noble WC. Compliment activation by Cornybacterium acnes. Br J Dermatol. 1978;98:583–584.
54. Webster GF, Leyden JJ, Norman ME, et al. Compliment activation in acne vulgaris: In vitro studies with Proprionibacterium acnes and Proprionibacterium granulosum. Infec Immun. 1978;22:523–529.
55. Burkhart CG, Cantrill J, Butcher CL, et al. Propionibacterium acnes: interaction with compliment and development of an enzyme-linked immunoassay for the detection of antibody. Int J Dermatol. 1999;38:200–203.
56. Millikan LE. The rationale for using a topical retinoid for inflammatory acne. Am J Clin Dermatol. 2003;4(2):75–80.
57. Czernielewski J, Michel S, Bouelier M, et al. Adapalene biochemistry and the evolution of a new topical retinoid for treatment of acne. J Eur Acad Dermatol Venereol. 2001; 15(suppl 3):5–12.
58. Gille J, Paxton LL, Lawley TJ, et al. Retinoic acid inhibits the regulated expression of vascular cell adhesion molecule-1 by cultured dermal microvascular endothelial cells. J Clin Invest. 1997;99(3):492–500.
59. Fumarulo R, Conese M, Riccardi S. Retinoids inhibit the respiratory burst and degranulation of stimulated human polymorphonuclear leukocytes. Agents and Actions. 1991;34:339–344.
60. Shroot B, Michel S. Pharmacology and chemistry of adapalene. J Am Acad Dermatol. 1997;36(6 pt 2):S96–S103.
61. Choi J-E, Piao MS, Lee J-B, et al. Propionibacterium acnes stimulates pro-matrix metalloproteinase-2 expression through tumor necrosis factor-a in human dermal fibroblasts. J Invest Dermatol. 2008;128:846–854.
62. Wauben-Penris PJJ, Cerneus DP, van de Hoven WE, et al. Immunomodulatory effects of tretinoin in combination with clindamycin. J Eur Acad Dermatol Venereol. 1998;11(S1): S2–S7.
63. Thiellitz A, Abdel-Naser MB, Fluhr JW, et al. Topical retinoids in acne—an evidence-based overview. J Dtsch Dermatol Ges. 2008;6(12):1023–1031. Epub 2008 May 13.
64. Wolf JE Jr. Potential anti-inflammatory effects of topical retinoids and retinoid analogues. Adv Ther. 2002;19:109–118.
65. Jones DA. The potential immunomodulatory effects of topical retinoids. Dermatology Online Journal. 2005;11(1):3.
66. Bikowski JB. Mechanisms of the comedolytic and anti-inflammatory properties of topical retinoids. J Drugs Dermatol. 2005;4(1):41–47.
67. Lavker RM, Leyden JJ, Thorne EG. An ultrastructural study of the effects of topical tretinoin on microcomedomes. Clin Ther. 1992;14:773–780.
68. Hirata N, Hiramatsu K, Hishi K, et al. Pretreatment of mice with clindamycin improves survival of endotoxic shock by modulating the release of inflammatory cytokines. Antimicrob Agents Chemother. 2001;45(9):2638–2642.
69. Akdeniz N, Ozbek CH, Metin A. Anti-inflammatory effects of tretinoin (all-trans-retinoic acid) 0.1% and adapalene 0.1% in rats. Clin Exper Dermatol. 2005;30:570–572.
70. Duffield JS. The inflammatory macrophage: a story of Jekyll and Hyde. Clinical Studies. 2003;104:27–38.
71. Dillehay DL, Walia AS, Lamon EW. Effects of retinoids on macrophage function and IL-1 activity. J Leukoc Biol. 1988;44:353–360.
72. Hensby C, Cacey D, Bouelier M, et al. The in vivo and in vitro anti-inflammatory activity of CD271: a new retinoid-like modulator of cell differentiation. Agents Actions. 1990;29:56–58.
73. Varani J, Jones J, Dame M, et al. Effects of all-trans retinoic acid on neutrophil-mediated endothelial cell injury in vitro and immune complex injury in rats. Am J Pathology. 1991;139(4):901–909.
74. Bécherel P-A, Mossalai MD, Goff LL. Mechanism of anti-inflammatory action of retinoids on keratinocytes. Lancet. 1994;344(8936):1570–1571.
75. Bécherel P-A, Le Goff L, Ktorza S, et al. CD23-Mediated nitric oxide synthase pathway induction in human keratinocytes is inhibited by retinoic acid derivatives. J Invest Dermatol. 1996;106:1182–1186.
76. Nagy I, Pivarcsi A, Koreck A, et al. Distinct strains of Propionibacterium acnes induce selective human b-defensin-2 and interleukin-8 expression in human keratinocytes through toll-like receptors. J Invest Dermatol. 2005;124:931–938.
77. Nagy I, Pivarcsi A, Kis K, et al. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infec. 2006;8:2195–2205.
78. Kurokawa I, Danby F, Ju Q, et al. New developments in our understanding of acne pathogenesis and treatment. Exp Dermatol. 2009;18:821–832.
79. Celerier P, Litoux P, Dreno B. In vitro modulation of epidermal inflammatory cytokines (IL-1, IL-6, TNF-a) by minocycline. Arch Dermatol Res. 1996;268:411–414.
80. Kang BY, Chung SW, Kim SH, et al. Retinoid-mediated inhibition of interleukin-12 production in mouse macrophages suppresses TH1 cytokine profile in CD4+ T cells. Br J Pharmacol. 2000;130:581–586.
81. Mouser PE, Baker BS, Seaton ED, et al. Propionibacterium acnes-reactive T helper-1 cells in the skin of patients with acne vulgaris. Letters to the Editor. J Invest Dermatol. 2003;121(5):1226–1227.
82. Griffiths CE, Voorhees JJ, Nickoloff BJ. Characterization of intracellular adhesion molecule-1 and HLA-DR expression in normal and inflamed skin: Modulation by recombinant gamma and tumor necrosis factor. J Am Acad Dermatol. 1989;20:617–629.
83. Leeuwenberg JF, von Asmuth EJ, Jeunhomme TM, et al. IFN-gamma regulates the expression of the adhesion molecule ELAM-1 and IL-6 production by human endothelial cells in vitro. J Immunol. 1990;145:2110–2114.
84. Bray MA. Retinoids are potent inhibitors of the generation of rat leukocyte leukotriene B4-like activity in vitro. Eur J Pharmacol. 1984;98(1):61–67.
85. Holland DB, Roberts SG, Cunliffe WJ. Localization of keratin 17 mRNA in acne. Sebaceous gland, acne and related disorders. J Invest Dermatol. 1997;108(abstr.):384.
86. Vega B, Jonard A, Michel S. Regulation of human monocyte toll-like receptor 2 (TLR2) expression by adapalene [abstract]. J Eur Acad Dermatol Venereol. 2002;16(Suppl 1):123–124.
87. Kampmann E, Johann S, van Neerven S, et al. Anti-inflammatory effect of retinoic acid on prostaglandin synthesis in cultured cortical astrocytes. J Neurochem. 2008;106:320–332.
88. Jalian HR, Liu PT, Kanchanapoomi M, et al. All-trans retinoic acid shifts Propionibacterium acnes-induced matrix degradation expression profile toward matrix preservation in human monocytes. J Invest Dermatol. 2008;128(12): 2777–2782. Epub 2008 Jun 19.
89. Del Rosso JQ, Schmidt NF. A review of the anti-inflammatory properties of clindamycin in the treatment of acne vulgaris. Cutis. 2010;85:15–24.
90. Richter JR, Forstrom LR, Kiistala UO, et al. Efficacy of the fixed 1.2% clindamycin phosphate , 0.025% tretinoin gel formulation (Velac) and a proprietary 0.025% tretinoin gel formulation (Aberela) in the topical control of facial acne. J Eur Acad Venereol. 1998;11:227–233.
91. Zouboulis CC, Derumeaux L, Decroix J, et al. A multicentre, single blind, randomized comparison of a fixed clindamycin phosphate/tretinoin gel formation (Velac) applied once daily and a clindamycin lotion formulation (Dalacin T) applied twice daily in the topical treatment of acne vulgaris. Br J Dermatol. 2000;143:498–505.
92. Rietschel R, Duncan BS. Clindamycin phosphate used in combination with tretinoin in the treatment of acne. Int J Dermatol. 1983;22:42–43.
93. Dreno B. Topical antibacterial therapy for acne vulgaris. Drugs. 2004:64(21):2389–2397.
94. Richter JRA, Bonsema MT, De Boulle KLVM, et al. Efficacy of a fixed clindamycin phosphate 1.2% tretinoin 0.025% gel formulation (Velac) in the topical control of facial acne lesions. J Dermatol Treat. 1998;9:81–90.
95. Leyden JJ, Krochmal L, Yaroshinsky A. Two randomized, double-blind, controlled trials of 2219 subjects to compare the combination clindamycin/tretinoin hydrogel with each agent alone and vehicle for the treatment of acne vulgaris. J Am Acad Dermatol. 2006;54:73–81.
96. Yaroshinsky A, Leyden JJ. The safety and efficacy of clindamycin (1%), as clindamycin phosphate and tretinoin (0.025%) for the treatment of acne vulgaris: a combined analysis of results from six controlled safety and efficacy trials conducted in Europe [abstract]. J Am Acad Dermatol. 2004;50(3 Suppl):P23.