NEW SECTION!
Skin Structure and Function: Translation of Research to Patient Care
Section Editors: Whitney High, MD; James Q. Del Rosso, DO; Jacquelyn Levin, DO
James Q. Del Rosso, DO, FAOCD; Jacquelyn Levin, DO
Dr. Del Rosso is Dermatology Residency Program Director, Valley Hospital Medical Center, Las Vegas, Nevada; Clinical Professor (Dermatology), Touro University College of Osteopathic Medicine, Henderson, Nevada; Las Vegas Skin & Cancer Clinics, Dermatology and Cutaneous Surgery, Las Vegas and Henderson, Nevada. Dr. Levin is PGY-2 (Dermatology), Largo Medical Center, Largo, Florida. Disclosure: Dr. Del Rosso serves as a consultant, researcher, and/or speaker for Allergan, Coria/Valeant, Galderma, Graceway, Intendis/Bayer, LeoPharma, Medicis, Onset Dermatologics, Ortho Dermatology, Pharmaderm, Promius, Ranbaxy, TriaBeauty, Unilever, and Warner-Chilcott. Dr. Levin reports no relevant conflicts of interest.
Abstract
It has been recognized for approximately 50 years that the stratum corneum exhibits biological properties that contribute directly to maintaining and sustaining healthy skin. Continued basic science and clinical research coupled with keen clinical observation has led to more recent recognition and general acceptance that the stratum corneum completes many vital “barrier” tasks, including but not limited to regulating epidermal water content and the magnitude of water loss; mitigating exogenous oxidants that can damage components of skin via an innate antioxidant system; preventing or limiting cutaneous infection via multiple antimicrobial peptides; responding via innate immune mechanisms to “cutaneous invaders” of many origins, including microbes, true allergens, and other antigens; and protecting its neighboring cutaneous cells and structures that lie beneath from damaging effects of ultraviolet radiation. Additionally, specific abnormalities of the stratum corneum are associated with the clinical expression of certain disease states. This article provides a thorough “primer” for the clinician, reviewing the multiple normal homeostatic functions of the stratum corneum and the cutaneous challenges that arise when individual functions of this thin yet very active epidermal layer are compromised by exogenous and/or endogenous factors.
Introduction
“In my paper of 1964, which established that the horny layer was a tissue made up of corneocytes, I could not have dreamed of the spectacular advances that have been brought to light by an international school of corneobiologists. I did not go any further than asserting that the stratum was a cellular barrier, the end product of a viable epidermis whose raison d’etre was to produce the dead stratum corneum…I did not have the vision to foresee that the stratum corneum would become very much alive.”
—Albert M. Kligman, MD
The quotation above by Dr. Albert Kligman, which he so brilliantly wrote in a textbook chapter entitled “A Brief History of How the Stratum Corneum Became Alive,” summarizes an important turning point in the history of dermatological research.[1] A brilliant discovery alone, regardless of how potentially revolutionary it may become, sits idle in a meaningless void unless its relevance is recognized and pursued with tenacity. Fortunately, the science of the stratum corneum (SC) has advanced exponentially due to the vision and dedication of a collection of leaders in the field of dermatology, referred to by Dr. Kligman as “corneobiologists.” Several researchers and clinicians over the past 5 to 6 decades have contributed to a wide body of published knowledge that supports our current understanding of how the SC, once thought to be biologically inert, actively contributes to the physiological homeostasis of skin.[1–3] Additionally, research continues to uncover specific abnormalities of the SC that contribute to certain dermatological diseases and/or impaired epidermal functions, and how some topical and systemic therapies may adversely affect SC integrity and function.[3]
This article reviews the following several subjects: 1) the history of SC science, 2) the homeostatic mechanisms whereby the SC functions to maintain the structural and functional integrity of skin, 3) impairments of or changes within the SC that relate to specific dermatological disorders, 4) endogenous and exogenous factors that create SC dysfunctions, and 5) our current understanding of clinical approaches to reverse or mitigate SC abnormalities resulting in improved therapeutic outcomes. It is important to recognize that “corneobiology,” and its many subsets of major clinical relevance, is in its infancy. Advancements in this field are undoubtedly part of a major groundswell as more information becomes available from basic science research. It is readily apparent how this information has already been incorporated into improved development of skin care products and better approaches to management of healthy and disease-affected skin.
Early Milestones in the History of Stratum Corneum
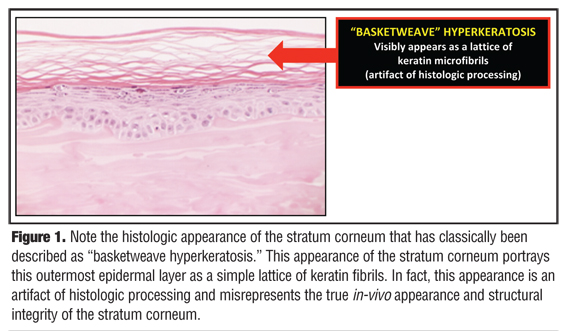
It was not too long ago that the established belief in dermatology was that the SC is “a graveyard of insoluble keratin fibrils” that collectively represent only the lifeless skeleton of what were previously keratinocytes that had completed their upward journey through the layers of the epidermis prior to their final destiny of desquamation.[1,2] In the 1950s, the SC, or “horny layer,” was described as the end product of living epidermal cells that disintegrated as they moved up in the process of squamous differentiation, ultimately becoming an “amorphous mass lacking a cellular structure.”[2,3] The concept that the SC is essentially a dead layer devoid of any functional activity was further entrenched in mainstream thinking by its perfunctory description as “basketweave hyperkeratosis” when viewed histologically after routine processing (Figure 1).[1,2]
{kind=link}
The belief that the stratum corneum was simply the “dead” outermost layer of skin, devoid of biological activity and function, persisted for several years. So how and when were the multiple talents of the SC discovered? The ability to harvest the SC intact and separate it from the remainder of the epidermis led to the belief that the SC was a homogenous SaranTM Wrap-like film that served primarily a protective function.[1,2]
“Perhaps no tissue is so physically maligned by processing for…microscopy…as is the stratum corneum…No tissue of crtitical importance for survival has been so intellectually maligned as well.”
—Peter M. Elias, MD
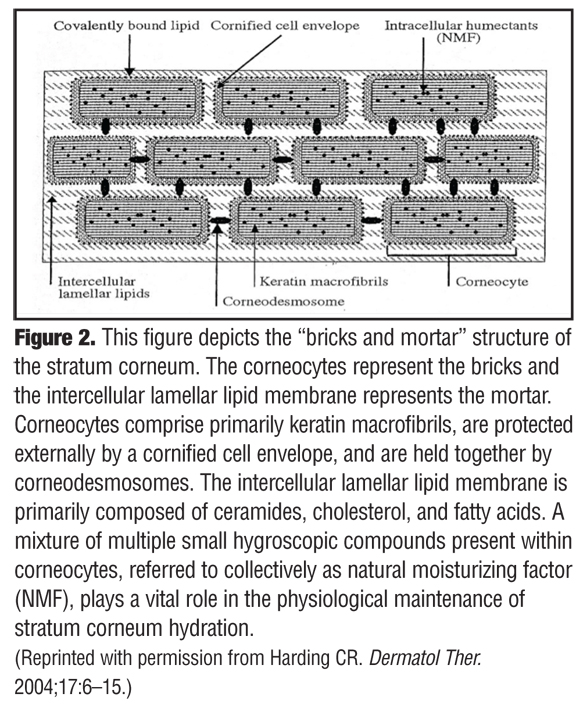
The above quotation by Dr. Peter Elias, which is from a book chapter he authored entitled, “The Epidermal Permeability Barrier: From SaranTM Wrap to Biosensor,” succinctly depicts how the significance of the SC was overlooked for decades.[2] Subsequent research and many publications over time led to the progressive recognition of the many physiological functions that occur within the SC, but widespread acceptance of this concept or its clinical relevance did not occur quickly.[3–20] One landmark publication that proved to be a major milestone in the history of relevant SC science was the seminal paper by Dr. Albert Kligman entitled, “Biology of the Stratum Corneum,” which was published in 1964.[2] This publication gave the previously ignored SC the respect it had long deserved by refuting prevailing recycled dogma with new information regarding SC structure. Up until this time, the strongly held prevailing belief described the SC as a simple, biologically inactive, outer epidermal layer comprising a fibrillar lattice of dead keratin or the nonviable final product of keratinocyte degradation that immediately precedes normal desquamation.[1,2] In fact, it was noted that the SC is made up of a collection of sturdy cellular-like structures, with an individual structure being termed a “corneocyte.” In time, the concept of the stratum corneum being viewed as “bricks” and “mortar” emerged, with the corneocytes representing the bricks and the intercellular lamellar lipid membrane representing the mortar between the bricks (Figure 2).[6]
{kind=link}
The bricks and mortar model of the SC, although it allows for initial conceptualization of the two-compartment macrostructure of the SC, is far too simplistic and does little to relate the constant sequence of varied functional activities that go on within this outermost epidermal layer, which measures just 10 to 20µm in thickness.[6,22] These diverse yet integrated functions of the SC collectively serve to detect, protect, respond, and/or adapt against several exogenous factors. Common exogenous insults to skin include exposure to irritants, allergens, and microbial organisms; climatic changes, especially those which cause low ambient humidity; acute and chronic photodamage; and iatrogenic insults, such as SC abnormalities associated with certain topical or oral medications. In many cases, the iatrogenic subset of exogenous insults is easily overlooked or not considered by the clinician, except where clinical relevance has been emphasized academically or is well-recognized in practice (i.e., the early irritant changes secondary to topical retinoid application). A separate and very relevant category is the endogenous factors, a spectrum of SC abnormalities that are inherently associated with specific disease states, such as atopic dermatitis, rosacea, and psoriasis. Importantly, SC impairments innately associated with specific underlying disease states do not allow for complete reversal of full SC function as some underlying disorders impart a baseline level of SC impairment even during periods of disease remission.[3,5,7,10,19,26]
Major Structural Components of the Stratum Corneum
The structural components of the SC are depicted in Figure 2. Corneocytes (bricks) comprised primarily of keratin macrofibrils, are protected externally by a cornified cell envelope, and are cohesively held together by corneodesmosomes.6 The cornified cell envelope is composed predominantly of proteins (e.g., loricin, involucrin) and a covalently bound outer lipid monolayer that is primarily made up of long chain ceramides.[6,22–24] Referred to as the “rivets” of the SC, corneodesmosomes serve to anchor the corneocytes within the SC and are composed of three major specialized proteins (desmoglein-1, desmocollin-1, and corneodesmosin). The primary function of the corneodesmosome is to maintain cohesive force between adjacent corneocytes until which time these rivets are degraded by water-dependent proteolytic enzymes involved in physiological desquamation.[6,22–25]
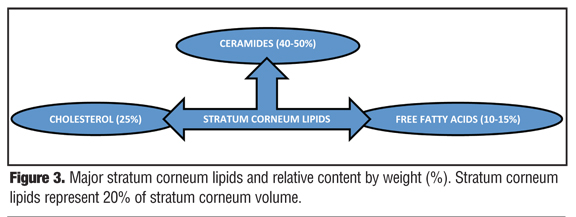
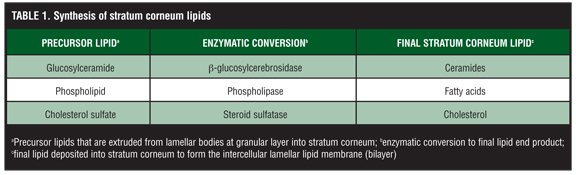
The intercellular lamellar lipid membrane comprises three major classes of lipid components present in a relative ratio of approximately 3:1:1 based on SC lipid content (% by weight): ceramides (40–50%; multiple subfractions), cholesterol (25%), and fatty acids (10–15%) (Figure 3). These major physiological SC lipids are produced enzymatically within the SC from specific precursor lipids (Table 1).[3,5,6,18,19,22] These precursor lipids are derived from lamellar bodies (LBs) within the granular layer, namely glycosylceramides, sphingomyelin, and phospholipids and are extruded from LBs along with antimicrobial peptides (Figure 3). Upon entry into the SC after extrusion from the LBs, these precursor lipids are enzymatically converted to ceramides 1–7, ceramides 2 and 5, and free fatty acids, respectively.[6,19,22] These two major hydrophobic lipid components of the SC, along with cholesterol, the third major component, and small quantities of other lipids, form a bilayer that comprises the intercellular lamellar lipid membrane.[3,5,22] Collectively, the physiological properties, relative composition, and the specialized intercellular compartmentalization of these physiological lipids within the intercellular lamellar lipid membrane serve to sustain SC water content, regulate water flux, and modify the rate and magnitude of transepidermal water loss (TEWL), all dynamic mechanisms that continuously function to maintain homeostasis.[5,22,26] As additional information about the SC and its varied functions continue to emerge, it is readily apparent that the SC is not a fixed, nondynamic, nonpliable wall, and its lipid composition is not random and inactive. Rather, the stratum corneum is a factory that is in operation at all times, is always in multi-task mode, and incorporates an array of both adaptive and protective “barrier” properties that are functionally dynamic and continual processes.[1–3,22]
{kind=link}
{kind=link}
What is Meant by the Term “Epidermal Barrier”?
When people use the term “epidermal barrier,” they are almost always referring to the ability of the SC to regulate TEWL, retain moisture for proper enzymatic desquamation, and provide selective permeability of exogenous and endogenous substances.[2,6,22,27] However, these functions are components of just one of the many “barrier” responsibilities that are carried out continuously by the SC, that is, the epidermal permeability barrier. In fact, the SC is multitalented as evidenced by its inherent ability to provide several other barrier properties. Examples include: 1) immunity barrier induced through specific receptor types, cell types, cytokines and chemokines with response via innate and/or cellular immune response patterns; 2) antioxidant barrier which protects against damaging effects of reactive oxygen species (ROS) via superoxide dismutase and other systems; 3) antimicrobial barrier via antimicrobial peptides (AMPs), such as cathelicidins and defensins within the lipid membrane, AMPs in sweat, and AMPs in sebum, and some SC lipids; 4) photoprotection barrier via both light reflectance and ultraviolet (UV) light photoprotectant properties of melanin and other chemicals; and 5) hormone receptor functions, such as peroxisome proliferator-activated receptors (PPARs) and liver X receptors (LXRs).[1–3,6,22,28–30] These different barrier functions of the SC are complexly intertwined with positive and negative feedback loops and biosensors that detect homeostatic abnormalities and stimulate self-repair mechanisms.[1–3,6,22,31–39]
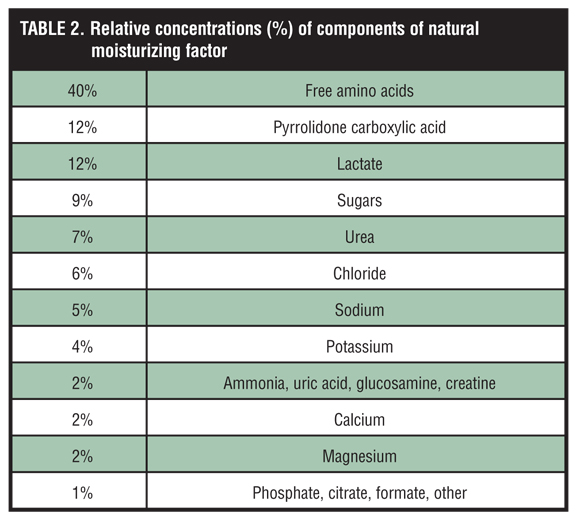
The ability of the SC to quickly adapt and initiate natural physiological recovery of the permeability barrier (self-repair) is demonstrated by the early detection of even modest increases in TEWL, followed by immediate release of lipids stored within existing lamellar bodies, which produces partial reversal of TEWL within minutes, and the marked increase within 2 to 3 hours of precursor lipid production in lamellar bodies that leads to formation of the major SC lipids.[6,22,35,38–39] These replenished SC lipids help to restore the functional integrity of the intercellular lamellar lipid membrane and provide some innate water-binding capacity within the intercellular spaces of the SC.[6,22,35,38,39] Additionally, intracellular SC humectancy and hydration is augmented by an upregulation in filaggrin production and its subsequent conversion into multiple degradation products within the granular layer.[32,34–37] The pivotal step of filaggrin degradation produces free amino acids, pyrrolidone carboxylic acid (PCA), and urocanic acid, which collectively with simple sugars and electrolytes ultimately form a unique hygroscopic “moisturizer” called natural moisturizing factor (NMF), the natural humectant present within corneocytes (Table 2).[1–3,6,37,40]
{kind=link}
Although much of our current knowledge and emphasis of the diverse barrier properties of the SC focus on the permeability barrier and its clinical relevance, a thorough understanding of the multiple complex interactions and feedback loops of the SC overall and recognizing specific abnormalities in the SC that contribute to unhealthy skin or diseased skin opens a door of opportunity to optimize SC repair by designing novel therapeutic approaches that target certain abnormalities or deficiencies.[1,3-6,14,15,18,19,22]
The next steps in appreciating the multiple roles of the SC in maintaining the functional health and integrity of both normal and disease-affected skin is to develop a thorough overall understanding of SC formation, its major functional components, and its adaptive physiology in healthy skin.
Formation of the Stratum Corneum
The SC represents the most superficial and final layer of maturation of the epidermis. However, where does the journey of the epidermis begin? The sequence of events occurs as keratinocytes traverse upward after their formation through the different layers (stratum) of the epidermis (Figure 4). Once arriving at a specialized zone of transition, the stratum granulosum, keratinocytes are sequentially modified and then converted to corneocytes, which comprise the SC prior to a final destiny of desquamation. The sequence of epidermal transition and the corresponding layers are 1) cellular proliferation with formation of keratinocytes within the basal layer (stratum basale); 2) keratinocyte squamous differentiation within the spinous layer (stratum spinosum); 3) formation of SC through transition of keratinocytes to corneocytes within the granular layer (statum granulosum) and the compacting layer (stratum compactum); 4) SC maturation with formation of the permeability barrier as corneocytes interdigitate with proper spatial relationship and cohesion in coordination with the intercellular lamellar lipid membrane (stratum corneum); 5) enzymatic corneocyte separation (stratum disjunctum); and 6) desquamation (into the atmosphere).[6,18,31,41,42]
{kind=link}
Importantly, the time course from the start of the journey at the basal cell layer to the endpoint of cerneocyte desquamation is generally estimated to take approximately four weeks, with the turnover of the SC being approximately half this duration. This is clinically relevant as therapies directed at modifying visible abnormalities within the SC (e.g., pitted keratolysis) may exhibit a lag time before improvement is apparent. This occurs as new “nondiseased” SC must first be formed before replacing previously “diseased” SC.
During the upward migration of epidermal cells, a stepwise sequence of active biological processes occurs that involves synthesis of specific structural protein, lipids, and enzymes; formation of structures integral to SC function; and activation of key enzymes, such as proteases, lipases, and transglutaminases, all of which impact directly upon SC health and functional integrity.[6,18,31,41–43] Basal keratinocytes of the stratum basale synthesize keratin filaments, adhesion molecules that attach the epidermis to the dermis, interkeratinocyte adhesion molecules, and cytokines and growth factors that regulate the proliferation and differentiation of the epidermis. After upward progression into the stratum spinosum, postmitotic keratinocytes continue the synthesis of keratin filaments, produce LBs composed of precursor lipids (glucosyl ceramides, phospholipids, sphingolipids, cholesterol sulfate), and form precursor proteins of the cornified envelope, which is comprised predominantly of loricrin (70%), and also contains other proteins (involucrin, cornifin, elafin, type II keratins, filaggrin, desmoglein, envoplakin, and small proline-rich proteins [SPRs]).[23,24,31,44] On the exterior surface of the cornified envelope surrounding each corneocyte, ceramides ultimately play an additional role in SC structural integrity as omego-hydroxyceramides form the corneocyte-lipid envelope, which osmotically maintains contents within the corneocyte and contributes to SC adhesion and lamellar organization. Once the synthesis of these multiple structural proteins and lipid precursors is completed, the keratinocytes are then prepared to move upward into the stratum granulosum, the zone of transition into the SC.
In the stratum granulosum, keratinocytes undergo a major “makeover” in preparing to become corneocytes. Within this layer, keratinocytes become flattened and contain specific organelles that serve to prepare the SC in maintaining the stability of the permeability barrier and other homeostatic functions. These organelles include keratohyalin granules, which contain profilaggrin, and LBs, which contain precursor lipids, certain protease enzymes, and AMPs. In the stratum granulosum, the LBs are positioned at the apical surface where they are in a “ready position” for exocytosis of contents into the lipid phase of the SC, which is the next layer of progression in the epidermal cell journey. However, an exception occurs in palmoplantar skin, where there is first cellular progression through an additional layer, the stratum lucidum, before becoming the SC.
Several major biological events occur upon transition from the stratum granulosum to the SC. At this point, keratinocytes become corneocytes after lysozomal degradation of their organelles. Also, there is extrusion by exocytosis of precursor lipids from the LBs into the lipid phase of the SC where they are subsequently enzymatically transformed into fatty acids and ceramides (Figure 4). These fatty acids and ceramides are then incorporated into the intercellular lamellar lipid membrane. Inside the corneocytes, profilaggrin, which originated from within the keratohyalin granules, is converted enzymatically to the protein filaggrin. Filaggrin subsequently migrates toward the periphery of the corneocyte and intermingles with keratin filaments to form the filament-matrix complex. On the surface of the corneocytes, calcium-dependent transglutaminases crosslink the proteins of the cornified cell envelope developing highly insoluble gamma glutamyl-lysine bonds. In the SC, the corneocytes are tightly adhered to each other by the cross-linked extracellular cornified envelope, desmosomal remnants, and the intracellular filament-matrix complex. Ultimately, a healthy SC continually performs several protective and adaptive physiological functions including mechanical shear and impact resistance, regulation of water flux and hydration, resistance to microbial proliferation and invasion, capacity to initiate inflammation via cytokine activation or dendritic cell activity, and selective permeability with exclusion of toxins, irritants, and allergens.[1,22,44]
Understanding the Complexities of the Stratum Corneum
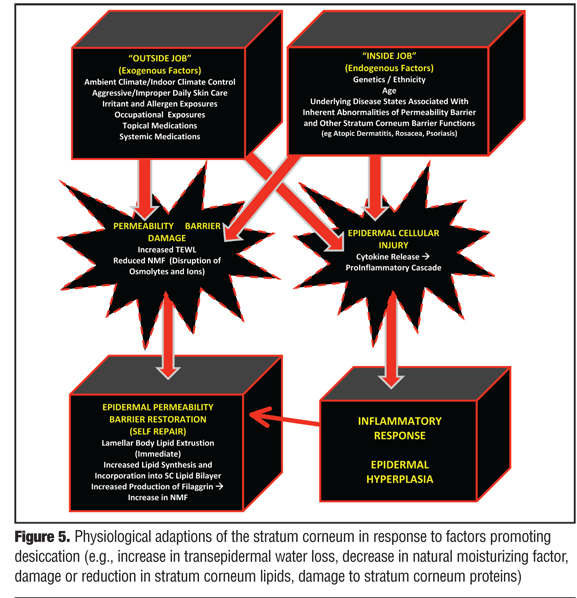
The first step in understanding the complexities and interactions among the structural components and adaptive physiological functions of the SC is to focus first on the permeability barrier and responses that occur within healthy or “normal” skin. Through maintenance of both proper skin hydration and resistance to shearing forces, the structural integrity of the SC is sustained from day to day, thus allowing other “barrier responsibilities,” such as immune surveillance and response, antimicrobial functions, and antioxidant activities to proceed. The ability of the SC to detect subtle changes in its own hydration status, that is, function as its own biosensor, is critical to the maintenance of its functional integrity and the overall appearance of healthy skin.[1–3,6,22] A variety of exogenous (“outside job”) and endogenous (“inside job”) factors, or both in combination, can contribute to altering the hydration status and integrity of the SC, as reviewed in more detail later.* Nevertheless, when there is either acute insult to the SC or chronic factors that increase TEWL and promote SC desiccation, the SC incorporates several adaptive mechanisms to restore its structural and functional integrity (Figure 5).[1–3,6,18,19,22,31–36,40–45]
{kind=link}
Once perturbed, the loss of permeability barrier function and subsequent increase in TEWL initiates multiple signaling cascades directed at restoring the permeability barrier of the SC. The major initial and quick response to an acute change in permeability barrier function is a temporary increase in LB secretion of precursor lipids into the SC where they are converted to the major lipids incorporated into the intercellular lamellar lipid membrane (lipid bilayer), and an increase in the biosynthesis of all major epidermal lipids which serves to sustain further self-repair.[22,35] The rapid response of release of LB lipids that were held in reserve can recover approximately 20 percent of the overall SC permeability barrier function.[22,35] Further repair requires addition synthesis of major SC lipids as well as other functions as discussed above.
In cases of chronic or frequently repetitive damage to SC protein and/or lipid components, more severe and more prolonged disruption of the SC permeability barrier occur simultaneously and work in concert adversely. The chronicity of this aberrant process results in an amplified signaling cascade that engages not only the desired epidermal homeostatic responses, but may also trigger the amplification of inflammatory events. In such cases, greater depth of epidermal and endothelial involvement occurs, which is more inflammatory in magnitude and can produce epidermal hyperplasia with chronic stimulation—both factors that are likely to play important roles in sustaining inflammatory dermatoses.[35,44,45] Protracted cases may be expressed clinically as severely xerotic and hyperkeratotic epidermal changes, especially on the distal fingers, distal toes, palms, soles, and heels.
The “Overstressed” Stratum Corneum: The Additive Effects of Endogenous and Exogenous Factors
Endogenous factors that adversely affect the epidermal permeability barrier also diminish the capacity of the SC to physiologically adapt to exogenous factors that disrupt permeability barrier function and increase TEWL. In other words, an individual cannot change his or her genetics, ethnicity, age, or underlying disease states that are inherently associated with permeability barrier impairment. As a result, these affected individuals are at an innate disadvantage when exogenous stresses that are placed upon the structural proteins and lipids of their SC exceed its restorative abilities. In such cases, the self-repair functions of the SC are not able to keep up with the speed and/or magnitude of permeability barrier restoration needed to fully reverse the excess in TEWL. Unless corrected by proper therapeutic intervention, the clinical sequelae of this scenario is initial progression to subclinical effects of SC desiccation, then to signs and symptoms of xerosis (dry skin), then to signs and symptoms of a flare of eczematous dermatitis, and finally to chronic eczematous and hyperkeratotic changes, all of which are discussed later.[3–18,22,26,32,37,38]
What endogenous characteristics have been identified that can retard the restorative capacities of the SC when exposed to exogenous stresses that disrupt the permeability barrier? Deficiencies and/or alterations in SC lipid content have been correlated with impairment of permeability barrier function, an important predisposing factor for development of xerosis.[3,5–8,22,26,38,42] A global decrease in epidermal lipid synthesis and SC lipids has been found in aged as opposed to younger skin.[38] A preliminary study, although small (N=71), has suggested that SC lipid composition and relative content may vary among different ethnicities living in the same environment.[46] With regard to underlying disease states, reduction in total SC lipid content, predominantly certain ceramide subfractions, is well documented in atopic dermatitis (AD), with the greatest decreases noted in actively eczematous skin; importantly, reductions in ceramides, although lower than in actively eczematous skin, have also been shown in xerotic and normal-appearing skin of patients with AD.[3,5-8,17,19,26,38,39,47] The inherent abnormalities of the SC permeability barrier in AD, associated with decreased SC lipid composition and content, and also in some cases filaggrin gene mutations, has been suggested as a major pathogenic factor in the development of the overall atopic diathesis, including allergic respiratory diseases and hypersensitivity (outside-in theory).[7–9,32,37,48,49]
What can be done to assist the overstressed SC in restoring the structural and functional integrity of the permeability barrier? As mentioned above, although the SC has the ability to self-repair a disrupted permeability barrier, there are many factors that can interplay to overstress the ability of the SC to fully adapt, especially when these factors are multiple, coexistent, repetitive, prolonged, and innate to other underlying medical disorders. A few active steps can be taken to optimize the health of the SC. Reducing exposure to and/or discontinuing practices that can cause damage to proteins and lipids of the SC is critical in decreasing the exogenous stress load on the permeability barrier. Such practices to avoid include overwashing; use of poorly formulated or harsh cleansers or true soaps (the latter due to alkalinity); use of cleansers with abrasives, exfoliants, or additives that are irritants; use of astringents; and use of topical medications that are not well formulated to reduce irritation potential.[4,6,40,42]
Appropriate skin care utilizing a well-formulated gentle cleanser and moisturizer, or barrier repair cream, has been shown to be beneficial in the management of disease states where the SC permeability barrier is impaired inherently by the disorder, its treatment, or both.[11–15,18,40,42,50–58] Many established moisturizers on the market today, in addition to incorporating conventional hydrating components, such as humectants, occlusives, and emollients, also include special additives or disease-specific ingredients, such as certain lipids, physiological humectants (i.e., hyaluronic acid), niacinamide, antioxidants, and amino acids.[4,15,40,50] Several of these formulations have been shown to markedly improve clinical signs and symptoms of AD and correct TEWL and corneometry parameters, thus demonstrating their ability to improve SC permeability barrier function.[51–53,55–58] Another study demonstrated improvement in recovery of the SC permeability barrier in chronically aged skin with use of a physiological lipid-based moisturizer.[56] Additional studies are warranted to further evaluate individual formulations, including attempts to differentiate properties that are relevant to clinicians and patients. Important differentiation parameters include several clinical performance measurements, regression analyses, and substantivity of effects. However, clinical differentiation does not stand alone. Proper nonbiased assessments of patient satisfaction are equally significant, including subjective feedback on therapeutic outcomes and physical formulation properties that influence adherence with continued use (i.e., texture; spreadability; ease of use; absence of malador; product “feel” such as greasy, tacky, sticky, etc).
More Information on Specific Components of the Stratum Corneum Permeability Barrier
As discussed earlier, NMF is a mixture of multiple water-soluble small hygroscopic compounds that act as natural humectants. The humectant properties of NMF are essential for the activity of proteases that regulate corneocyte desquamation and moisture retention.[18,42] Free amino acids comprise approximately 40 percent of NMF and may be derived from multiple sources, including eccrine sweat, degradation of structural SC proteins, such as desmosomes and hemidesmosomes, citrulline originating from hair follicles, and degradation of filaggrin.[40,59] However, the release and degradation of filaggrin from keratohyaline granules of the stratum granulosum is believed to be the predominant source of free amino acids and PCA in the SC, with quantitative analyses showing that between 70 and 100 percent of the total free amino acids of the SC are derived from histidine-rich proteins.[59]
As stated previously, the keratohyalin granules and profilaggrin first appear in the granular layer of the epidermis. Profilaggrin in the keratohyalin granule is converted to filaggrin via enzymatic proteolysis as the cells of the granular layer transition into the SC layer. In this transition, the keratohyaline granules also intermix with keratin filaments forming the filament-matrix complex. Filaggrin is degraded into several free amino acids (histidine, glutamine, arginine), PCA, urocanic acid, ornithine, citrulline, and aspartic acid. These degradation products exhibit functions within the permeability barrier of the SC, including maintenance of hydration, pH (acidity), buffering capacity of the SC, and physiological desquamation, all of which contribute to healthy skin function and appearance.[1–3,6,22,32–34,36,37,40,42,59,60]
The clinical relevance of filaggrin and its proper degradation in helping to maintain the functional integrity of the SC permeability barrier is strongly supported by the link between filaggrin loss-of-function gene mutations and SC-impaired disorders, such as icthyosis vulgaris (IV) and AD, both of which are chronically xerotic and associated with inherent basal cutaneous pH readings and impaired buffering capacity.[32,37,61]
Why is Maintenance of Stratum Corneum Hydration so Important?
There are three main factors that maintain an optimal level of hydration of the epidermis and the SC.[18,22,31,40–44] The first is the formation and packaging of epidermal lipids and their organization into the intercellular lamellar lipid membrane in the SC (lipid bilayer). In the bilayer, the lipids are organized in an orthorhombic gel phase to effectively impede the passage of water through the intercellular space. Also, long-chain ceramides form a lipid layer on the surface of the cornified cell envelope. The second is the interdigitation of corneocytes within the intercellular lamellar lipid membrane, which creates an indirect and longer diffusion path for water as it attempts to traverse the SC, thus retarding TEWL. The third is the presence of NMF in the corneocytes of the SC, formed predominantly during the degradation of filaggrin.
The structural and functional integrity of the SC is highly dependent on adequate water content as many of the enzymes that catalyze vital SC functions are hydrolytic and do not occur efficiently if water is present below a threshold concentration.[22,40,42] In addition to permeability characteristics, water serves to plasticize the SC as water content directly affects other physical properties, such as flexibility and pliability, resistance to shearing forces, individual desquamation of corneocytes, skin pH, and formation of the cornified cell envelope.[1,2,6,18,23,24,31,40,42–44,59,60,62]
The delicate interplay between SC water content and proper physiological SC function is further exemplified by the observation that the hydration level within the SC is not uniform under healthy physiological conditions in normal skin, ranging from approximately 15 to 25 percent at the skin surface, to approximately 40 percent at the juncture of transition between the granular layer and the SC, to about 70 percent within the lower portion of the epidermis below the SC.[20,44,62] It is believed that this water gradient is established in part by a discontinuity in the water-binding capacities between different corneocyte cell layers and may also reflect the need for different hydration levels to support specific enzymatic functions at certain depth locations within the epidermis. On electron microscopy, the corneocytes within SC levels of low water content appear less swollen than the corneocytes that have more water content at different depth points, as the swelling of corneocytes is directly proportional to skin hydration.[18]
Ultimately, in properly hydrated skin, the corneocytes shed as individual invisible cells, as the integrity of the epidermis and SC is physiologically sound from bottom to top. From a clinical standpoint, properly hydrated skin, reflected by a properly hydrated and minimally damaged SC, appears healthy, is pliable, and devoid of scaling or dryness. When the SC is desiccated, the degradation of corneodesmosomes by hydrolytic enzymes is impaired, and corneocytes tend to “clump” before shedding, thus causing roughness with visible scaling and flaking. The skin appears scaly, rough, and dull in appearance, as the features of xerotic skin emerge.[1,4,13,17,18,25,40,42,63]
What is Xerosis (Dry Skin)?
Regardless of the cause, dry skin, also referred to as xerosis, is the visible reaction pattern that occurs when TEWL becomes excessive and exceeds the ability of SC to adequately repair the permeability barrier. Although the term “dry skin” is commonly used by consumers, cosmeticians, scientists, researchers, physicians, and other healthcare professionals to describe one of the most common human afflictions, a clear definition has been elusive.[63] Ultimately, dry skin is best defined clinically by its presenting signs and symptoms. Dry skin is dull in appearance, rough, scaly, flakey, and tight, often with associated pruritus, and in some cases discomfort especially in low ambient humidity.[63]
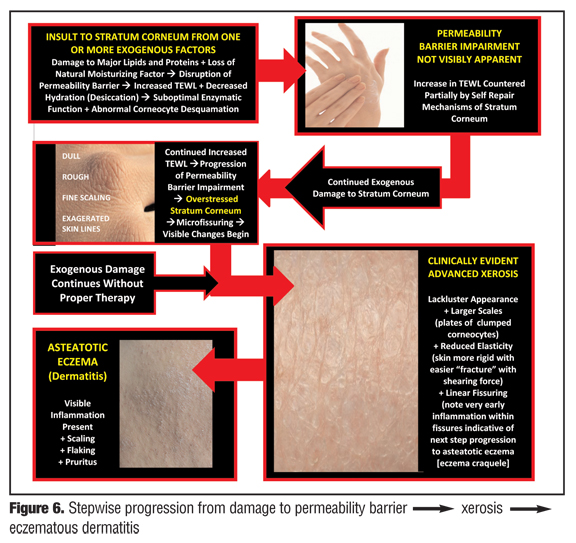
The progression from normal skin to clinically evident xerosis and subsequent eczematous changes occurs in a stepwise fashion (Figure 6). Insult to the SC, usually from one or more exogenous factors (e.g., overwashing; overbathing; use of harsh skin cleansers, astringents, or exfoliants; exposure to irritants; occupational exposures; climatic changes; low ambient humidity; certain topical medications or cosmeceuticals) causes damage and loss (“stripping”) of SC lipids, damage to important SC proteins, and loss of components of NMF, leading to increased TEWL and desiccation.[4,6,40,42,63] With continued SC damage that is not countered by proper therapeutic intervention, the SC becomes “overstressed,” that is, the continued insults causing disruption of the permeability barrier exceed the reparative capabilities of the SC. Over time, the clinical presentation of xerotic changes become more severe and exaggerated, and as a result of cytokine release and stimulation of an inflammatory cascade (Figure 5), can progress to eczematous dermatitis (e.g., eczema craquele, asteatotic eczema, nummular eczema, irritant hand eczema, exacerbation of AD) with associated pruritus and erythema. Epidermal hyperplasia with formation of hyperkeratotic eczema, which is associated with little or no visible inflammation, can also result from chronic damage to the permeability barrier on the hands and feet, often with associated deep fissures and pain, especially on the fingertips.
{kind=link}
One of the most common exogenous factors that can cause epidermal damage is overwashing or overbathing, especially when combined with use of poorly formulated or harsh cleansers, especially true soaps, which are alkaline. Use of a poorly formulated, harsh, or irritant cleanser can induce deleterious effects on the SC, depending on formulation characteristics and specific ingredients (e.g., type of surfactant).[4,11–13,16,17,40,42,58,63,64] Strong cleansers that are highly efficient in removing dirt, oil, and other debris from the skin surface are also likely to produce the greatest magnitude of damage to the permeability barrier of the SC through stripping lipids and components of NMF and/or damage to SC proteins. Recognition of the importance of mild cleansers in the management of skin disease and in the maintenance of the SC permeability barrier has spurred the development of advanced mild cleanser formulations incorporating technologies and ingredients that provide a beneficial balance between gentle cleansing and efficient cleaning. These advanced cleanser systems induce negligible damage to the SC and in some cases actually deposit lipid into the SC after cleansing and rinsing.[13,40,42,58,64,65]
Certain topical products, including over-the-counter (OTC) skin care products with certain additives (e.g., alpha-hydroxy acids, astringents, abrasive granules, retinol) can induce damage to the SC leading to changes related to SC desiccation and cutaneous irritation. Topical prescription medications intended to treat specific skin disorders or cosmetic concerns may actually cause or exacerbate damage to the SC permeability barrier and possibly other epidermal functions depending on the pharmacological properties of the active ingredient(s). As will be discussed later, examples of medications that may adversely affect the SC include topical corticosteroids, topical retinoids, and benzoyl peroxide. However, the extent to which a topical formulation affects the SC permeability barrier may vary somewhat depending on the ingredients used in the vehicle. The vehicles of some products inherently induce SC damage due to specific inert ingredients used in the formulation, independent of the active ingredient(s) of the product. Other vehicles are designed to mitigate SC damage that is associated with the active ingredient(s) it contains, thus reducing potential for adverse effects related to SC desiccation. The formulation details including vehicle ingredients, information on SC-related parameters (e.g., TEWL, corneometry), and the cutaneous tolerability data of each product is important in understanding the potential effects that a product may have on the SC permeability barrier and other functions. Systemic agents that can impair the SC permeability barrier are the HMG-CoA reductase inhibitor agents used to lower serum cholesterol (e.g., simvastatin, atorvastatin, lovastatin, pravastatin). These lipid-lowering agents, commonly referred to as “the statins,” interfere with cholesterol synthesis, with cholesterol being a major component of the SC intercellular lamellar lipid membrane, and may also alter the cornified cell envelope.[66]
What Other Factors Can Affect the Permeability Barrier of the Stratum Corneum?
The pH of the SC has a significant influence on the barrier functions of the SC, with alterations in pH alone enough to cause clinically relevant changes. For example, the neutral to alkaline nature of the SC in the early neonatal period affects SC permeability and desquamation and facilitates the growth of opportunistic organisms such as Candida albicans, creating a milieu that is supportive of diaper dermatitis over approximately the first three months of life.[44] After that point, the SC pH develops its “sweet spot” for optimal physiological and homeostatic function and integrity, which is an acidic pH commonly referred to as the “acid mantle” of the skin.
The acidic nature of the human skin surface develops early in life, within the first few months, and is believed to be influenced by both exogenous and endogenous sources.[43,44,67,68] Exogenous sources of an acidic pH include free fatty acids produced by lipases from normal microbial flora, pilosebaceous-derived free fatty acids, and lactic acid and other eccrine-derived products.[44] Endogenous factors that produce a physiological acidic pH of the SC include cis-urocanic acid produced by degradation of histidine, free fatty acid production from phospholipids catalyzed by secretory phospholipase A2 (degradation of LB phospholipids), and via sodium-proton exchange mechanisms.[44] Acidification of skin pH has been shown to normalize SC integrity and permeability barrier recovery in neonates with neutral skin pH; expedite SC epidermal barrier recovery; optimize activity of lipid-processing enzymes and processing of LB-derived precursor lipids; and regulate corneocyte desquamation, cohesion, and integrity.[44] In addition, acidic SC pH favors growth of normal bacterial flora, as opposed to an alkaline pH, which is more supportive of the growth of pathogens such as Staphylococcus aureus and C. albicans.[67]
Other factors that can influence the functional and/or structural integrity of the SC include maintenance of a normal epidermal calcium gradient and regulation of epidermal development and SC permeability barrier homeostasis by nuclear hormone receptors and their ligands.[44]
What are the Other Major “Barrier Responsibilities” of the Stratum Corneum Beyond the Permeability Barrier?
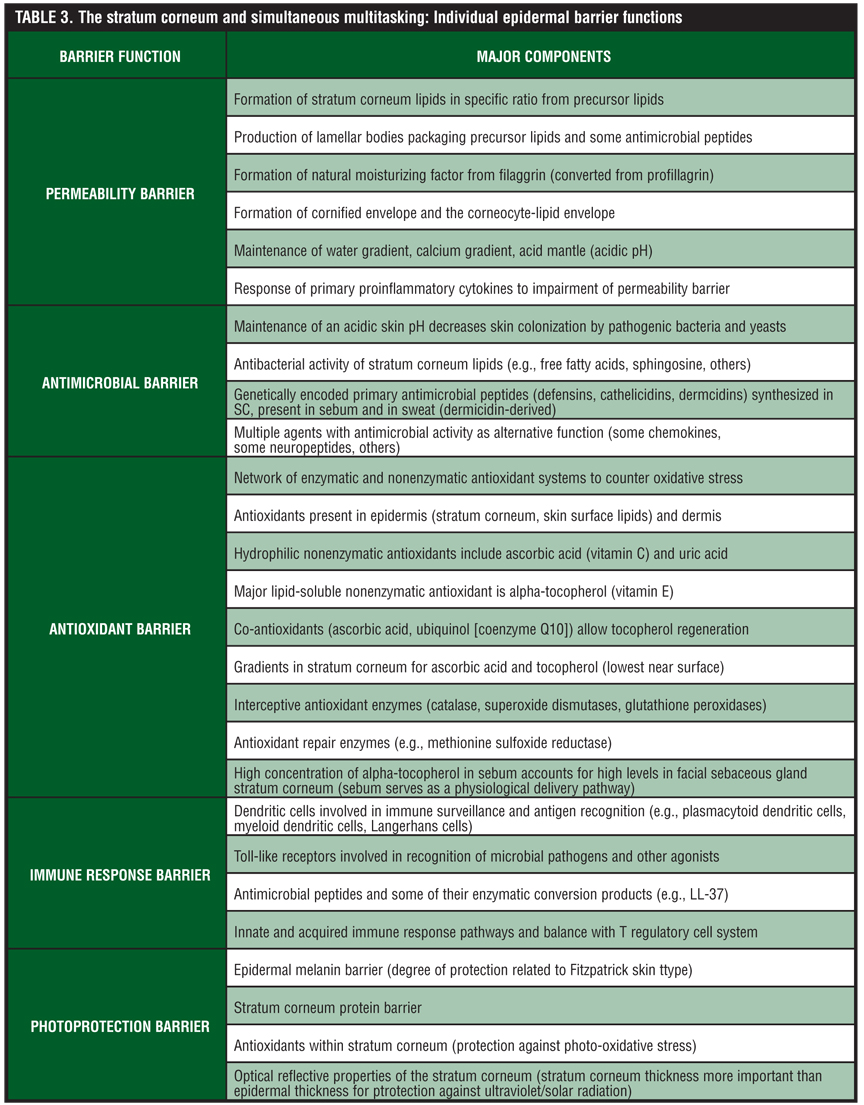
Although discussion of the SC permeability barrier is the focal point of this paper, there are several other “barrier responsibilities” related to specific components and functions of the SC (Table 3). A major responsibility of the SC is the antimicrobial barrier, which serves to provide protection against invasion and infection by microbial organisms, including bacteria, fungi, and viruses, but also is innately involved with some pathways of cutaneous inflammation.[28–30,69–72] The antimicrobial barrier comprises several AMPs, which are generally small, cationic polypeptides with the capacity to directly inhibit the growth of microbial organisms or indirectly mitigate microbial invasion by activating host immune response pathways. Thus, AMPs often serve as mediators of inflammation that affect epithelial and inflammatory cells, cellular proliferation, wound healing, cytokine and chemokine production, and chemotaxis.[69–72]
{kind=link}
While cathelicidins and defensins comprise the major families of cutaneous AMPs, there are a variety of AMPs that have been identified, including some found in sebum and sweat.[28–30,69–72] While the upper stratum spinosum and stratum granulosum are the primary sites where AMPs are synthesized, these molecules are subsequently delivered into the SC where their functional activity occurs. Some AMPS, such as human beta defensin-2 (HBD-2) and cathelicidins have been shown to be localized within the LBs, which deliver these AMPs to the SC along with precursor lipids.[28,69]
In disorders where there is compromise of the SC permeability barrier and LBs are deficient, there is often associated dysfunction in the antimicrobial barrier of the SC. Examples of disorders where alterations in AMPs have been noted include AD and psoriasis.[5,6,44,73] The expression of HBD-2 and LL-37, the latter a product of cathelicidin processing, are downregulated in AD. These findings help to explain the high susceptibility of AD skin colonization and infection with S. aureus, and the predisposition to viral infections associated with AD, such as molluscum contagiosum and eczema herpeticum, the latter related to a decrease in the cathelicidin-derived protein LL-37 in AD.[74,75] As LL-37 exhibits antiviral properties, a decrease in LL-37 accounts at least partially for the predisposition to viral infections that often affect patients with AD. In addition, an increase in primary epidermal pro-inflammatory cytokines (e.g., tumor necrosis factor [TNF], interleukin-1[IL-1], interleukin-6 [IL-6]) in response to epidermal insult and disruption of the SC permeability barrier is believed to assist in self-repair. However, prolonged or frequently repetitive damage to the SC can produce adverse clinical effects secondary to chronic inflammation and epidermal hyperproliferation.[76]
Other barrier functions of the SC are depicted in Table 3. Importantly, although the systems involved in each barrier function are unique and often complex, the interplay between these “functional units” of the SC is critical to the overall health and appearance of the skin. Loss of function in any one area can correlate with clinically apparent changes within the skin, and in some cases specific disease states.[6,44]
In some cases, the underlying dysfunction of the SC may be genetically programmed, such as with AD or the ichthyoses, while in other cases the underlying cause may be primarily iatrogenic, self-induced, or associated with acquired disease states. The epidermal antioxidant barrier incorporates a network of multiple nonenzymatic and enzymatic systems, which protect against oxidative changes induced by multiple exogenous causes.[77] Examples of such exogenous causes include chemical oxidants, ultraviolet radiation, air pollutants, microbial organisms including bacteria and fungi, and phagocyte-generated ROS that are directed at clearing pathogenic microbial organisms but also produce collateral damage to cutaneous tissues. Lastly, the cutaneous photoprotection barrier is provided by four potential mechanisms—the epidermal melanin barrier, the SC protein barrier, SC antioxidants, and optical reflective properties of the SC (direct correlation with SC thickness).[77]
Disease-Affected Skin and Associated Stratum Corneum Abnormalities
Atopic dermatitis. The “poster disease” that has popularized the clinical significance of “epidermal barrier dysfunction” is AD. AD is a complex cutaneous disorder associated with several operative pathogenic factors that interact to create a varied spectrum of phenotypes seen every day in clinical practice by dermatologists, allergists, and primary care physicians.[78] These multiple pathogenic factors include genetics (especially parental history); environmental influences, such as air pollutants, indoor exposures (e.g., house mite antigens, pet-related antigens), lifestyle (urban versus rural), dietary exposures, effect of antibiotic use in childhood on maturation of immunity; innate abnormalities of the epidermis including the SC permeability barrier (e.g., decrease in ceramide subfractions, filaggrin gene mutations, impaired ability to sustain SC hydration), the SC antimicrobial barrier (e.g., reduced levels of some AMPs, decrease in LL-37), and the immune response barrier; dysregulation of both innate and acquired immune response with alteration in chemokines and cytokines (e.g., Th2 imbalance, Th2- Th1 shift, increase in IgE, eosinophilia); hyper-responsive to both cutaneous allergens and irritants; role of microbial organisms (e.g., S. aureus colonization and superantigen production); and neurogenic and neuroimmunological factors (e.g., neuropeptides and neurotropins in epidermal nerve fibers near mast cells and Langerhans cells, increased blood levels of nerve growth factor, and substance P directly correlating with disease activity).[3,5,6–9,32,37,38,43,44,47–49,69,78] The presence of multiple pathogenetic factors in AD confounds the ability to differentiate the relative significance of each with regard to clinical presentation and therapeutic management, both in the AD population overall, and in the individual patient. Nevertheless, some of these factors are more available to clinicians as “low hanging fruit,” that is, they are easier to address in the treatment of AD with what is currently available, including measures to counter the innate impairment of the SC permeability barrier and to decrease cutaneous S. aureus colonization.
From a clinical perspective, it is estimated that 45 percent of patients with AD present within the first six months of life, 60 percent within the first 12 months of life, and 85 percent before five years of age.[78] The extrinsic subtype of AD, which represents 60 to 90 percent of cases, is also associated with polyvalent IgE sensitization to inhalant and/or food allergens, explaining subsequent development of asthma, seasonal rhinitis, and sometimes food hypersensitivity in many patients with AD.[79] Although AD is most predominant in childhood and adolescence, and is typified by intermittent flares of eczematous skin changes and marked pruritus often involving multiple skin sites, persistence into adulthood is not uncommon, with many cases presenting as recurrent forms of eczema.[80–83] These include localized forms of eczema that are often recurrent, such as lichen simplex chronicus (LSC, “neurodermatitis”), hand eczema, eyelid dermatitis, genital pruritus (e.g., vulvar pruritus, vulvar hyperplastic dystrophy, scrotal pruritus often with LSC), asteatotic dermatitis on the legs, and sometimes diffuse forms of adult AD presenting as xerotic-pruritic skin without visible eczema, nummular eczema, and susceptibility to winter itch.
Xerosis, flares of eczematous dermatitis, and pruritus, are consistent clinical features of AD.[80–84] Additionally, S. aureus colonization is a very common association that is often clinically silent or can present as crusted eczema (“impetiginization,” infectious eczematoid dermatitis) or the actual presence of secondary staphylococcal infection.[80–85] Importantly, S. aureus colonization has been implicated in the pathogenesis of AD, playing a role in initiation of and/or prolongation of flares of active eczema through superantigen production and stimulation of a subsequent inflammatory cascade.[50–56,85]
SC lipids. There is a plethora of data documenting the significance of epidermal “barrier” disruptions in AD, inclusive of impairment of the SC permeability barrier, SC antimicrobial barrier, and the immune response barrier.[3,5–9,29–32,37–44,47–49,60,69–72,74,75] Abnormalities in SC lipids have been confirmed in eczematous skin, in xerotic skin, and in uninvolved skin in patients with AD.[3,5,6,8,17,26,38,39,42–44,47,48] The greatest decrease ceramide subfractions in AD patients has been shown with ceramide-1 (CER-1) in both lesional (eczematous) and nonlesional skin (visibly uninvolved without presence of eczema); however, ceramide-2 through ceramide-6 (CER-2–CER-6) were also markedly decreased in both lesional and nonlesional skin.[3,5,26,39] Decreases in CER-1 and total ceramide content were also found in the SC of AD patients with clinically xerotic skin that was otherwise devoid of visible eczematous dermatitis.[3,7] Correlation of SC ceramide content with SC permeability barrier function in patients with AD demonstrated a marked decrease in the amounts of CER-1 and CER-3, and a direct correlation of reduction in CER-3 with an increase in TEWL.[3,5,47] A three- to five-fold increase in TEWL has been observed in lesional skin of AD patients as compared to nonlesional skin.3,5,86,87 Up to a two-fold increase in TEWL has been noted in clinically normal skin and xerotic skin (without presence of eczematous changes) in AD patients who present with active flares of eczema at other cutaneous sites.[3,5,86,87]
Filaggrin story. As discussed earlier, the role of profilaggrin production, its conversion to filaggrin, and the degradation of filaggrin to form major components of NMF are vital to physiological maintenance of SC hydration and the functional integrity of the SC permeability barrier.[6,31,32,34,37,42,88,89] Loss-of-function mutations in the filaggrin gene are collectively the strongest and most widely replicated genetic risk factor for developing AD.[88] It is estimated that as many as half of all AD cases are accounted for by a defect in at least one filaggrin gene null allele.89 In other cases of AD, an acquired impairment of filaggrin may be present, likely the result of downregulated filaggrin protein expression induced by interleukin-4 (IL-4) and interleukin-13 (IL-13), which are increased in patients with AD.[89] Interestingly, it has been suggested that characteristics of filaggrin genes may be associated with the dry skin phenotype in the overall population, unrelated to association with specific underlying disease states such as AD. Specifically, predisposition to dry skin may be related to genetically determined “repeat copy units” in the polypeptide that is encoded for by the filaggrin gene.[88,89]
The development of filaggrin and its degradation products is a delicate balance of production, proteolysis, and inhibition of the process.89 Every intricate step of this process needs to be functioning and in place in order to produce a healthy functional SC permeability barrier, as any alteration in the process can lead to permeability barrier dysfunction. Although loss-of-function filaggrin gene mutations are of major significance in the pathophysiology of AD, at least in many patients, a defect/deficiency in filaggrin alone may not be enough to cause AD, as suggested by a study completed in filaggrin-deficient mice, and also as some patients with filaggrin gene mutations present with ichthyosis vulgaris alone without AD.[88–90] In the murine filaggrin-deficient skin study, AD-like skin changes were induced only after intentional exposure to allergens, suggesting that antigen transfer through a defective SC permeability barrier may also be a key mechanism underlying elevated IgE allergic sensitization and initiation of cutaneous inflammation in humans with filaggrin-related atopic disease.[90]
Immune response. AD patients exhibit multiple abnormalities in their immune response barrier. A normally functioning innate immune response system recognizes pathogens and coordinates a response including anatomical and cellular mechanisms to combat invasion and infection.[6,28,30,69,71,72,75] In AD, there is both impaired microbial recognition and response.[44,69,78] Pattern recognition receptors (PRR), such as toll-like receptors (TLRs), recognize antigens and in response trigger cytokine release (e.g., IL-1, TNF), which initiates an immune response.[76,91] Unfortunately in AD, it has been shown that there are several specific TLR defects, including polymorphisms of TLR2, which may lead to a higher rate of S. aureus infection.[92] Another important player associated with impairment of the immune response barrier is CD14, a receptor responsible for recognizing lipopolysaccharides and other bacterial cell wall components. Low levels of CD14, similar to TLR-2 polymorphism, impair pathogen recognition and initiation of immune response in AD patients, with low CD14 levels in breast milk correlated with an increased susceptibility for development of AD in children.[93,94]
Antimicrobial peptides. Defensins, cathelicidins, and dermicidins are all AMPS that are diminished in AD.[44,69,91] As a result of reduced AMPs and other antimicrobial substances in AD, affected patients are more susceptible to bacterial colonization and infection, such as with S. aureus.[84,85,91,95] Additionally, levels of LL-37 and HBD-2 are lower in lesional skin of patients with AD, as compared to psoriasis lesions and normal skin, and the same has been shown with several other keratinocyte AMPs in lesional skin of AD versus psoriasis.[96,97] As noted earlier, LL-37, a product of enzymatic conversion of cathelicidins, has known antiviral activity (e.g., herpes simplex virus). Therefore, deficient levels or activity of LL-37 may explain the increased susceptibility of AD patients to certain viral infections, such as eczema herpeticum and possibly molluscum contagiosum.[98]
It has been shown that 90 percent of patients with AD exhibit colonization with S. aureus involving both lesional and nonlesional skin as compared to five percent of healthy controls, with up to 60 percent being toxin-producing strains of S. aureus.[91,99] In addition, decrease in HBD-2 and LL-37 in acute and chronic lesions of AD support susceptibility to S. aureus.[95] Additionally, a mutation in a TLR2 gene (R753Q) is found in increased frequency in AD, correlating with greater severity of disease, higher serum levels of IgE, and increased susceptibility to colonization with S. aureus.[100] There is evidence that S. aureus colonization plays a pathogenic role in AD, serving as to both trigger and prolong a flare of active eczema, through production of specific toxins called superantigens (e.g., enterotoxins A, B, and toxic shock syndrome toxin-1 [TSST-1]), which stimulate expanded populations on skin-homing T cells.[85] Evidence supporting the role of staphylococcal superantigens in AD includes the correlation of AD severity with presence of IgE antibodies to superantigens, superantigen-induced activation of infiltrating mononuclear cells augmenting allergen-induced inflammation, induction of mast cell degranulation, induction of skin-homing receptor on T cells by superantigens, and activation of Th2 cells by superantigens.[85] In addition to superantigens, staphylococci can produce nonsuperantigenic toxins, such as alpha-toxin, and compounds that can contribute to cutaneous inflammation, such as protein A and lipoteichoic acid.[101]
Topical corticosteroids. The effects of topical corticosteroids (TCS) on the SC will be discussed later under psoriasis.
Acne vulgaris. Disease state. There is little data confirming an innate impairment of the SC permeability barrier in acne vulgaris (AV). In one small study of patients with AV (N=36), increased TEWL, reduced SC hydration based on conductance testing, reduction in total ceramides, and reduction in free sphingosine (%) were noted in patients with AV as compared to controls.102 Further study is needed to evaluate baseline characteristics of the SC in untreated patients with AV.
Acne therapies. Some topical acne medications are associated with signs of cutaneous irritation, such as peeling, scaling, and erythema. As a result, the association of a topical formulation or ingredient with impairment of the SC permeability barrier or other SC functions is an important consideration. Cutaneous application of benzoyl peroxide has been shown to increase TEWL, to oxidize SC antioxidants (e.g., alpha tocopherol [vitamin E]), and to cause lipid peroxidation; however, supplementation with topical vitamin E did not correct the increase in TEWL, but did reduce markers of lipid peroxidation.[103] Topical retinoids commonly induce an initial period of “retinization,” characterized by peeling, flaking, and erythema (“retinoid dermatitis”), especially during the first few weeks after starting application. The use of a barrier-enhancing moisturizer prior to and during use of topical tretinoin facilitated adaptation to topical-retinoid induced signs of cutaneous irritation, decreased TEWL, and improved cutaneous hydration as measured by conductance.[104]
Rosacea. Skin sensitivity, characterized by associated signs and/or symptoms, are common in patients with the two most common clinical subtypes of rosacea, unrelated to use of any medical therapies.[10,14,58] In untreated adult patients with papulopustular rosacea (N=915) pooled from multiple studies, dryness, scaling, and edema were reported by 65 to 69 percent, 51 to 58 percent, and 32 to 38 percent of subjects, respectively.[14,104] In this same collective group of rosacea patients, facial skin burning, stinging, and pruritus were reported by 34 to 36 percent, 29 to 34 percent, and 49 to 52 percent of subjects, respectively.[105] According to a survey conducted by the National Rosacea Society in 1997 involving 1,023 patients with rosacea, 82 percent of these patients reported skin hyperirritability, burning, stinging, and sensitivity from commonly used skin care products.[106] Of the female responders, the three most common items reported to be irritating to facial skin included astringents/toners, soaps, and exfoliating agents. Of the male responders, the three most common causes of facial irritation included soap, cologne, and shaving lotion.
Impairment of the SC permeability barrier, as demonstrated by increased centrofacial TEWL has been documented in patients with erythematotelangiectatic rosacea (ETR), papulopustular rosacea (PPR), and also perioral dermatitis, and at least partially explains the inherent skin sensitivity that is so commonly expressed by patients with rosacea.10 Skin sensitivity in patients with rosacea, likely related to disease-inherent SC permeability barrier impairment, is further supported by the results of lactic acid sting testing, completed in patients with ETR (n=7), PPR (n=25), and healthy skin (n=32 controls).[107 ]In this study, a positive sting test reaction with 5% lactic acid was found in 100 percent of ETR patients, 68 percent of PPR patients, and 19 percent of control patients.
Psoriasis. Although the pathophysiology of psoriasis is multifactorial and related to predisposing gene variants, genetic modifiers, and environmental factors, SC abnormalities have been identified that may contribute to the pathogenesis of the disease.[73,108] SC abnormalities that can translate to impairment of SC barrier functions include abnormal disposition on LBs in some phenotypes, alterations in formation of the cornified cell envelope, and decrease in filaggrin expression.[73]
Psoriasis phenotype. In both erythrodermic psoriasis and actively developing plaque psoriasis, a large number of LBs are formed. However, many of the LBs remain “entombed” in the cytosol of corneocytes, resulting in defective delivery of SC lipids to the intercellular space where the lipid bilayer is formed.[73] However, in chronic plaque psoriasis, LB contents are formed and secreted into the intercellular space of the SC, which is more consistent with nonpsoriatic healthy skin. A decrease in CER-1 has been noted from psoriatic plaques as compared to uninvolved skin and controls, with direct correlation between the CER-1 level and the increase of TEWL.109 Interestingly, increase in TEWL noted in association with psoriasis is greatest in erythrodermic and actively developing plaque lesions, intermediate in magnitude in chronic plaque psoriasis lesions, and lowest in nonlesional skin.[110]
Differentiation markers. Markers of epidermal differentiation are also altered in psoriasis as compared to normal skin. These include an increase in keratinocyte transglutaminase type 1, which modulates an important step in the formation of the cornified cell envelope, decrease in filaggrin expression, increase in involucrin, increase in hyperproliferative keratins K6 and K16, and decrease in K1 and K10, which are markers of terminal differentiation.[73,111]
Antimicrobial barrier. In comparison to patients with AD, patients with psoriasis exhibit less susceptibility to infection. This is due to impairment of the antimicrobial barrier in AD, while an increase in AMPs has been noted in psoriasis.[95–97]
Topical corticosteroids. Application of TCS can result in visible skin abnormalities, such as atrophy, purpura, telangiectasias, and irreversible striae, which primarily result from dermal changes. In addition, microscopic and other structural changes have been associated with TCS use, many of which involve the SC and have barrier implications, such as epidermal atrophy; decreased microvasculature; decreased keratinocyte size; decreased ceramides, free fatty acids, and cholesterol; and increased TEWL.[112,113]
It has been demonstrated that as little as three days of application of topical clobetasol in both human and murine skin can induce subclinical adverse changes in the epidermis.[113] These effects include deterioration in epidermal barrier homeostasis characterized by delayed barrier recovery, abnormal SC integrity and cohesion, and the global inhibition of lipid synthesis. This study also showed improved barrier recovery and improvement in SC cohesion and integrity with the application of a physiological lipid-based moisturizer. These observations suggest that it may be prudent to use a well-formulated moisturizer as an adjunct to TCS therapy to ameliorate the adverse effects on the SC permeability barrier.
The adverse effects of TCS on the SC may be partially mitigated by the simultaneous use of a vitamin D analog based on a murine skin study. Topical calcitriol was shown to counteract the impairment of the epidermal permeability barrier and the antimicrobial barrier induced by TCS.[114] Specifically, the addition of topical calcitriol to TCS therapy significantly improved TEWL compared to TCS use alone (p=0.024), increased LB density compared to TCS use alone (p=0.031), and significantly upregulated major enzymes involved in lipid synthesis, HMG-CoA (p=0.02), fatty acid synthase (p=0.02), and serine-palmitoyl transferase (p<0.01). In addition, calcitriol was able to reverse the reduction of AMP expression induced by TCS, such as cathelicidin-related AMP (CRAMP) (p<0.01) and mouse beta-defensin 3 (mBd3) (p<0.01).
Management of Stratum Corneum Permeability Barrier Impairment
Maintenance of SC hydration, including assisting the SC in self-repair when conditions are adverse, is vital to sustaining healthy function and appearance of the skin. The use of a well-formulated gentle skin cleanser that produces negligible damage to the SC and a moisturizing product designed to sustain the integrity of the SC permeability barrier, with both facilitating and expediting physiological repair of the SC, is an optimal strategy for both healthy skin and disease-affected skin. Several studies have demonstrated that proper use of well-formulated skin care products augments therapeutic response to overall treatment in a variety of skin conditions, such as AD and rosacea, and/or reduces adverse effects associated with other therapies that adversely affect the SC.[4,6,11,16,18,22,35,40,42,50–58,64,65,104,115,116] Importantly, moisturizing agents that incorporate quality conventional ingredients, such as humectant and occlusive agents, contain physiological lipids or ingredients that augment skin lipid synthesis, and/or include other agents that facilitate permeability barrier restoration, may be an optimal choice as these formulations are designed to target specific SC abnormalities that lead to xerotic skin changes, at least in some cases. Agents that are predominantly occlusive and/or humectant may exhibit less substantivity, may produce less overall impact on physiological barrier restoration, and are often less likely to be preferred by patients for continued use due to a greasy or sticky texture.[22,42,35,50–55] Importantly, individual formulations must be evaluated based on their own performance in both research investigations and after careful observation during clinical use. Additional research is needed to further our understanding of optimal formulations, including differentiation and use for specific disease states.
Acknowledgment
*Dr. Del Rosso would like to credit Anthony Mancini, MD, a pediatric dermatologist in Chicago, Illinois. A few years ago during a lecture Dr. Mancini was presenting, Dr. Del Rosso heard for the first time the use of the terms “inside job” for endogenous factors, and “outside job” for exogenous factors, applied specifically to atopic dermatitis. Immediately after hearing this, Dr. Del Rosso incorporated Dr. Mancini’s description to elucidate these concepts more clearly and informed Dr. Mancini how much he liked these two descriptions from a teaching perspective.
References
1. Kligman AM. A brief history of how the dead stratum corneum became alive. In: Elias PM, Feingold KR, eds. Skin Barrier. New York: Taylor & Francis; 2006:15–24.
2. Elias PM. The epidermal permeability barrier: from Saran Wrap to biosensor. In: Elias PM, Feingold KR, eds. Skin Barrier. New York: Taylor & Francis; 2006:25–32.
3. Proksch E, Elias PM. Epidermal barrier in atopic dermatitis. In: Bieber T, Leung DYM, eds. Atopic Dermatitis. New York: Marcel Dekker; 2002:123–143.
4. Del Rosso JQ. Understanding skin cleansers and moisturizers: the correlation of formulation science with the art of clinical use. Cosmet Dermatol. 2003;16:19–31.
5. DiNardo A, Wertz PW. Atopic dermatitis. In: Leyden JJ, Rawlings AV, eds. Skin Moisturization, 1st ed. New York: Marcel Dekker; 2002:165–178.
6. Harding CR. The stratum corneum: structure and function in health and disease. Dermatol Ther. 2004;17:6–15.
7. Yamamoto A, Serizawa S, Ito M, et al. Stratum corneum abnormalities in atopic dermatitis. Arch Dermatol Res. 1991;283:219–223.
8. Cook M, Danby S, Vasilopoulos Y, et al. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol. 2009;129:1892–1908.
9. Ogawa H, Yoshiike TJ. A speculative view of atopic dermatitis: barrier dysfunction in pathogenesis. Dermatol Sci. 1993;5:197–204.
10. Dirschka T, Tronnier H, Folster-Holst R. Epithelial barrier function and atopic diathesis in rosacea and perioral dermatitis. Br J Dermatol. 2004;150:1136–1141.
11. Del Rosso JQ, Baum EW. Comprehensive medical management of rosacea: an interim study report and literature review. J Clin Aesthet Dermatol. 2008;1(1):20–25.
12. Draelos, ZD. Concepts in skin care maintenance. Cutis. 2005;76(6 Suppl):19–25.
13. Subramanyan K. Role of mild cleansing in the management of patient skin. Dermatol Ther. 2004;17(suppl 1):26–34.
14. Del Rosso JQ. The use of moisturizers as an integral component of topical therapy for rosacea: clinical results based on the assessment of skin characteristics study. Cutis. 2009;84:72–76.
15. Loden M. Role of topical emollients and moisturizers in the treatment of dry skin barrier disorders. Am J Clin Dermatol. 2003;4(11):771–778.
16. Misra M, Ananthapadmanabhan KP, Hoyberg K, et al. Correlation between surfactant-induced ultrastructural changes in epidermis and transepidermal water loss. J Soc Cosmet Chem. 1997;48:219–234.
17. Rawlings AW, Watkinson A, Rogers J, Mayo HJ, Scott IR. Abnormalities in stratum corneum structure, lipid composition, and desmosome degradation in soap-induced winter xerosis. J Soc Cosmet Chem. 1994;45:203–220.
18. Rawlings AV, Scott IR, Harding CR, Bowser PA. Stratum corneum moisturization at the molecular level. J Invest Dermatol. 1994;103(5):731–740.
19. Menon GK, Norlen L. Stratum corneum ceramides and their role in skin barrier function. In: Leyden J, Rawlings A, eds. Skin Moisturization. New York, NY: Marcel Dekker, Inc.; 2002:31–60.
20. Bouwstra JA, de Graaff A, Gooris GS, et al. Water distribution and related morphology in human stratum corneum at different hydration levels. J Invest Dermatol. 2003:120:750–758.
21. Kligman AM. Biology of the stratum corneum. In: Montagna W, ed. The Epidermis. New York: Academic Press Inc.; 1964.
22. Elias PM. Physiologic lipids for barrier repair in dermatology. In: Draelos ZD, ed. Cosmeceuticals, 1st ed. Philadelphia: Elsevier-Saunders; 2005:63–70.
23. Koch PJ, Roop DR, Zhou Z. Cornified envelope and corneocyte-lipid envelope. In: Elias PM, Feingold KR, eds. Skin Barrier. New York: Taylor & Francis; 2006:97–110.
24. Tetsuji H. Cornified envelope. In: Rawlings AV, Leyden JJ, eds. Skin Moisturization, 2nd ed. New York: Informa Healthcare; 2009:83–97.
25. Haftek M, Simon M, Serre G. Corneodesmosomes: pivotal actors in stratum corneum adhesion and desquamation. In: Elias PM, Feingold KR. Skin Barrier. New York: Taylor & Francis; 2006:171–189.
26. Imokawa G, Abe A, Higaki Y, et al. Decreased level of ceramides in stratum corneum of atopic dermatitis: an etiologic factor in atopic dry skin? J Invest Dermatol. 1991;96:523–526.
27. Elias PM, Feingold KR. Stratum corneum barrier function: definitions and broad concepts. In: Elias PM, Feingold KR. Skin Barrier. New York: Taylor & Francis; 2006:1–3.
28. Braff M, DiNardo A, Gallo R. Keratinocytes store the antimicrobial peptide cathelicidin in lamellar bodies. J Invest Dermatol. 2005;124:94–400.
29. Elias P. Stratum corneum defensive functions: an integrated view. J Invest Dermatol. 2005;5:183–200.
30. Schroder JM, Harder J. Antimicrobial skin peptides and proteins. Cell Mol Life Sci. 2006;63:469–486.
31. Elias PM, Feingold KM. Permeability barrier homeostasis. In: Elias PM, Feingold KR. Skin Barrier. New York: Taylor & Francis; 2006:337–361.
32. O’Regan GM, Irvine AD. The role of filaggrin in the atopic diathesis. Clin Exp Allergy. 2010:40:965–972.
33. Scott IR, Harding CR. Barrett JG. Histidine rich proteins of the keratohyaline granules: source of the free amino acids, urocanic acid and pyrolidone carboxylic acid in the SC. Biochem Biophys Acta. 1982:719;110–111.
34. Katagiri C, Sato J, Nomura J, et al. Changes in environmental humidity affect the water-holding property of the stratum corneum and its free amino acid content, and the expression of filaggrin in the epidermis of hairless mice. J Dermatol Sci. 2003;31:29–35.
35. Mao-Qiang M, Brown BE, Wu-Pong S, et al. Exogenous nonphysiologic vs. physiologic lipids: divergent mechanisms for correction of permeability barrier dysfunction. Arch Dermatol. 1995;131:809–816.
36. Elias PM, Ansel JC, Woods LD, et al. Signaling networks in barrier homeostasis: the mystery widens. Arch Dermatol. 1996;132:1505–1506.
37. McLean I. Loss-of-function mutations in the filaggrin gene lead to reduced level of natural moisturizing factor in the stratum corneum. J Invest Dermatol. 2008;128:2117–2119.
38. Choi MJ, Maibach HI. Role of ceramides in barrier function of healthy and diseased skin. Am J Clin Dermatol. 2005;6:215–223.
39. Imokawa G. Ceramides as natural moisturizing factors and their efficacy in dry skin. In: Leyden JJ, Rawlings AV, eds. Skin Moisturization, 1st ed. New York: Marcel Dekker; 2002:267–302.
40. Del Rosso JQ. Moisturizers: function, formulation, and clinical applications. In: Draelos ZD. Cosmeceuticals. Philadelphia: Saunders Elsevier; 2009:97–102.
41. James WD, Berger TG, Elston DM. Skin: basic structure and function. In: James WD, Berger TG, Elston DM, eds. Andrews’ Diseases of the Skin, 10th ed. Philadelphia: Saunders-Elsevier; 2006:1¬13.
42. Johnson AW. Cosmeceuticals: function and the skin barrier. In: Draelos ZD. Cosmeceuticals. Philadelphia: Saunders-Elsevier; 2009:7–14.
43. Cork MJ, Moustafa M, Danby S, et al. Skin barrier dysfunction in atopic dermatitis. In: Rawlings AV, Leyden JJ, eds. Skin Moisurization, 2nd ed. New York: Informa Health; 2009:211–239.
44. Fluhr JW, Darlenski R. Skin barrier. In: Revuz J, et al, eds. Life Threatening Dermatoses and Emergencies in Dermatology. Heidelberg, Germany: Springer-Verlag; 2009:3–18.
45. Elias PM, Woods LD, Feingold KR. Epidermal pathogenesis of inflammatory dermatoses. Am J Contact Dermat. 1999;10:119–126.
46. Jungersted JM, Hogh JK, Hellgren LI, et al. Ethnicity and stratum corneum ceramides. Br J Dermatol. 2010;163:1169–1173.
47. DiNardo A, Wetz P, Giannetti A, et al. Ceramide and cholesterol composition of the skin of patients with atopic dermatitis. Acta Derm Venereol. 1998;78:27–30.
48. Elias PM, Hatano Y, Williams ML. Basis for the barrier abnormality in atopic dermatitis: outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol. 2008;121: 1337–1343.
49. Leung DYM. Atopic dermatitis: the skin as a window into the pathogenesis of chronic allergic disease. J Allergy Clin Immunol. 1995;96: 302–319.
50. Rawlings AV, Canestrari DA, Dobkowski B. Moisturizer technology versus clinical performance. Dermatol Ther. 2004;17:49–56.
51. Chamlin SL, Kao J, Frieden IJ, et al. Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: changes in barrier function provide a sensitive indicator of disease activity. J Am Acad Dermatol. 2002;47:198–208.
52. Lucky AW, Leach AD, Laskarzewski P, et al. Use of an emollient as a steroid sparing agent in the treatment of mild to moderate atopic dermatitis in children. Pediatr Dermatol. 1997;14:321–324.
53. Hanifin JM, Hebert AA, Mays SR, et al. Effects of a low-potency corticosteroid formulation plus a moisturizing regimen in the treatment of atopic dermatitis. Curr Therapeutic Res. 1998;59:227–233.
54. Boguniewicz M. Conventional topical treatment of atopic dermatitis. In: Bieber T, Leung DYM, eds. Atopic Dermatitis. New York: Marcel Dekker; 2002:453–477.
55. Draelos ZD. The effect of ceramide-containing skin care products on eczema resolution duration. Cutis. 2008; 81:87–91.
56. Zettersten EM, Ghadially R, Feingold KR, et al. Optimal ratios of topical stratum corneum lipids improve barrier recovery in chronologically aged skin. J Am Acad Dermatol. 1997;37:403–408.
57. Frankel A, Sohn A, Patel RV, Lebwohl M. Bilateral comparison study of pimecrolimus 1% cream and a ceramide-hyaluronic acid emollient foam in the treatment of patients with atopic dermatitis. J Drugs Dermatol. 2011;10(6):666.
58. Levin J, Miller R. A guide to the ingredients and potential benefits of over-the-counter cleansers and moisturizers for rosacea patients. J Clin Aesthet Dermatol. 2011;4(8):31–49.
59. Levin J, Maibach H. Human buffering capacity considerations in the elderly. In: Farage M, Miller K, Maibach HI, eds. Textbook of Aging Skin. Berlin: Springer-Verlag; 2010:139–145.
60. Hachem JP, Man MQ, Crumrine D, et al. Sustained serine protease activity by prolonged increase in pH leads to degradation of lipid processing enzymes and profound alterations of barrier function and stratum corneum integrity. J Invest Dermatol. 2005;125:510–520.
61. Levin J, Maibach HI. Human skin buffering capacity: overview. In: Zhai H, Wilhelm KP, Maibach HI, eds. Marzulli and Maibach’s Dermatotoxicology, 7th ed. Boca Raton: CRC Press, Taylor and Francis Group; 2008:971–980.
62. Caspers PJ, Lucassen GW, Carter EA, et al. In-vivo confocal Raman microspectroscopy of the skin: noninvasive determination of molecular concentration profiles. J Invest Dermatol. 2001;116:434–442.
63. Rawlings AV, et al. Dry and xerotic skin conditions, In: Leyden JJ, Rawlings AV, eds. Skin Moisturization, 1st ed. New York: Marcel Dekker, Inc.; 2002:119–143.
64. Ananthapadmanabhan KP, Moore DJ, Subramanyan K, et al. Cleansing without compromise: the impact of cleansers on the skin barrier and the technology of mild cleanser. Dermatol Ther. 2004;17:16–25.
65. Ananthapadmanabhan KP, Yang L, Vincent C, et al. A new technology in mild and moisturizing cleansing liquids. Cosmetic Dermatol. 2009;22(6):307–316.
66. Sparsa A, Boulinguez S, Le Brun V, et al. Acquired ichthyosis with pravastatin. J Eur Acad Dermatol Venereol. 2007;21:549–550.
67. Schmid-Wendtner MH, Korting HC. The pH of the skin surface and its impact on barrier function. Skin Pharmacol Physiol. 2006;19:296–302.
68. Behne MJ. Epidermal pH. In: Rawlings AV, Leyden JJ, eds. Skin Moisturization, 2nd ed. New York: Informa Healthcare; 2009:125–148; 163–180.
69. DiNardo A, Gallo RL. Cutaneous barriers in defense against microbial invasion. In: Elias PM, Feingold KR. Skin Barrier. New York: Taylor & Francis; 2006:363–377.
70. Eissa A, Diamandis EP. Kallikrein-related peptidases: an emerging family of pivotal players in epidermal desquamation and barrier function. In: Rawlings AV, Leyden JJ, eds. Skin Moisturization, 2nd ed. New York: Informa Healthcare; 2009:125–148.
71. Lee SH, Jeong SK, Ahn SK. An update of the defensive barrier function of skin. Yonsei Medical Journal. 2006;47(3):293–306.
72. Nagy I, Kemeny L. Skin immune system. In: Revuz J, et al, eds. Life Threatening Dermatoses and Emergencies in Dermatology. Heidelberg, Germany: Springer-Verlag; 2009:19–28.
73. Ghadially R. Psoriasis and ichthyosis. In: Leyden JJ, Rawlings AV, eds. Skin Moisturization, 1st ed. New York: Marcel Dekker; 2002:165–178.
74. Frohm M, Agerberth B, Ahangari G, et al. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem. 1997;272:15258–15263.
75. Schauber J, Dorschner RA, Coda AB, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. 2007;117:803–811.
76. Biao L, Elias PM, Feingold KR. The role of the primary cytokines, TNK, IL-1, IL-6, in permeability barrier homeostasis. In: Elias PM, Feingold KR. Skin Barrier. New York: Taylor & Francis; 2006:305–318.
77. Thiele JJ. The epidermal antioxidant barrier. In: Elias PM, Feingold KR, eds. Skin Barrier. New York: Taylor & Francis; 2006:379–397.
78. Bieber T, Prolss J. Atopic dermatitis. In: Gaspari AA, Tyring SK, eds. Clinical and Basic Immunodermatology. London, Springer-Verlag; 2008:193–206.
79. Wuthrich B, Schmid-Grendelmeier P. Definition and diagnosis of intrinsic versus extrinsic atopic dermatitis. Conventional topical treatment of atopic dermatitis. In: Bieber T, Leung DYM, eds. Atopic Dermatitis. New York: Marcel Dekker; 2002:1–20.
80. Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol (Stockh). 1980;92(Suppl):44–47.
81. Diepgen TL, Fartsch M, Hornstein OP. Evaluation and relevance of atopic basic and minor features in patients with atopic dermatitis and in the general population. Acta Derm Venereol (Stockh). 1989;144:50–54.
82. Williams HC, Pembrokle AC, Burney PGF, et al. Community validation of the UK Working Party’s diagnostic criteria for atopic dermatitis. Br J Dermatol. 1996;135:12–17.
83. Schultz Larsen F, Diepgen T, Svensonn A. Clinical criteria in diagnosing AD: the Lillehammer criteria 1994. Acta Derm Venereol (Stockh). 1996;76(Suppl 196):115–119.
84. Leyden JJ, Marples RR, Kligman AM. Staphylococcal aureus in the lesions of atopic dermatitis. Br J Dermatol. 1974;90:525–530.
85. Leung DYM. The role of Staphylococcus aureus in atopic dermatitis. In: Bieber T, Leung DYM, Eds. Atopic Dermatitis, 1st ed. New York: Marcel Dekker; 2002:401–418.
86. Seidenari S, Giusti G. Objective measurement of the skin of children affected by atopic dermatitis: a study of pH, capacitance and TEWL in eczematous and clinically uninvolved skin. Acta Derm Venereol. 1995;75:429–433.
87. Schafer L, Kragballe K. Abnormalities in epidermal lipid metabolism in 11 patients with atopic dermatitis. J Invest Dermatol. 1991;96:10–15.
88. Irvine AD, McClean WHI. Breaking the (un)sound barrier: filaggrin is a major gene for atopic dermatitis. J Invest Dermatol. 2006;126:1200–1202.
89. Uitto J, McGrath JA. The role of fliaggrin in skin disease. In: Rawlings AV, Leyden JJ, eds. Skin Moisturization, 2nd ed. New York: Informa Health; 2009:57–67.
90. Fallon PG, Sasaki T, Sandilands A, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genetic. 2009;41:602–608.
91. Martin DB, Gaspari AA. Toll-like receptors In: Gaspari AA, Tyring SK, eds. Clinical and Basic Immunodermatology. London: Springer-Verlag; 2008:67–84.
92. Ahmad-Nejad P, Mrabet-Dahbi S, Breuer K, et al. The toll-like receptor 2 R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J All Clin Immunol. 2004;113:565–567.
93. Zdolsek HA, Jenmalm MC. Reduced levels of soluble CD14 in atopic children. Clin Exp Allergy. 2004:34:532–539.
94. Rothenbacher D, Weyermann M, Beermann C, Brenner H. Breastfeeding, soluble CD14 concentration in breast milk and risk of atopic dermatitis and asthma in early childhood: birth cohort study. Clin Exp Allergy. 2005:35;1014–1021.
95. Jalian HR, Kim J. Antimicrobial peptides. In: Gaspari AA, Tyring SK, eds. Clinical and Basic Immunodermatology. London: Springer-Verlag; 2008:131–145.
96. Ong PY, Ohtake T, Brandt C, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–1160.
97. de Jongh GJ, Zeeuwen PL, Kucharekova M, et al. High expression levels of keratinocyte antimicrobial peptides in psoriasis compared with atopic dermatitis. J Invest Dermatol. 2005;125(6):1163–1173.
98. Howell MD, Wollenberg A, Gallo RL, et al. Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Immunol. 2006;117(4):836–841.
99. Leung D. Infection in atopic dermatitis. Curr Opin Pediatr. 2003;15(4):399–404.
100. Werfel T, Heeg, K, Neumaier M, et al. R753Q polymorphism defines a subgroup of patients with atopic dermatitis having severe phenotype. J Allergy Clin Immunol. 2004;113(3):565–567.
101. Morath S, Geyer A, Hartung T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J Exp Med. 2001;193:393–398.
102. Yamamoto A, Takenouchi, Ito M. Impaired water barrier function in acne vulgaris. Arch Dermatol Res. 1985;287:214–218.
103. Weber ST, Thiele JJ, Han N, et al. Topical alpha-tocotrienol supplementation inhibits lipid peroxidation but fails to mitigate increased transepidermal water loss after benzoyl peroxide treatment to human skin. Free Radical Biol Med. 2003;34(2):170–176.
104. Draelos ZD, Ertel KD, Berge CA. Facilitating facial retinization through barrier improvement. Cutis. 2006;78:275–281.
105. Elewski BE, Fleischer AB, Pariser DM. A comparison of 15% azelaic acid gel and 0.75% metronidazole gel in the topical treatment of papulopustular rosacea. Arch Dermatol. 2003;139:1444–1450.
106. Torok HM. Rosacea skin care. Cutis. 2000;66(4 Suppl):14–16.
107. Lonne-Rahm SB, Fischer T, Berg M. Stinging and rosacea. Acta Derm Venereol (Stockh). 1999;79: 460–461.
108. Nestle FO. Psoriasis. In: Gaspari AA, Tyring SK, eds. Clinical and Basic Immunodermatology. London: Springer-Verlag; 2008:207–216.
109. Motta S. Abnormality of water barrier function in psoriasis: role of ceramide fractions. Arch Dermatol. 1994;130(4):452–456.
110. Ghadially R, Reed JT, Elias EM. Stratum corneum structure and function correlates with phenotype in psoriasis. J Invest Dermatol. 1996;107(4):558–564.
111. Schroeder WT. Type 1 keratinocyte transglutaminase: expression inn human skin and psoriasis. J Invest Dermatol. 1992;99(1):27–34.
112. Del Rosso J, Friedlander SF. Corticosteroids: options in the era of steroid-sparing therapy. J Am Acad Dermatol. 2005;53:850–858.
113. Kao JS, Fluhr JW, Man MQ, et al. Short-term glucocorticoid treatment compromises both permeability barrier homeostasis and stratum corneum integrity: inhibition of epidermal lipid synthesis accounts for functional abnormalities. J Invest Dermatol. 2003;120:456–464.
114. Hong SP, Oh Y, Jung M, et al. Topical calcitriol repairs epidermal permeability and antimicrobial barriers induced by corticosteroids. Br J Dermatol. 2010;162: 1251–1260.
115. Tabata N, O’Goshi K, Zhen YX, et al. Biophysical assessment of persistent effects of moisturizers after their daily applications: evaluation of corneotherapy. Dermatology. 2000;200:308–313.
116. Johnson AW. Overview: fundamental skin care-protecting the barrier. Dermatol Ther. 2004;17:1–5.