Grace K. Kim, DO; James Q. Del Rosso, DO, FAOCD
Dr. Kim is from Valley Hospital Medical Center, Las Vegas, Nevada. Dr. Del Rosso is Dermatology Residency Director, Valley Hospital Medical Center, Las Vegas, Nevada. Disclosure: Dr. Kim reports no relevant conflicts of interest. Dr. Del Rosso is a consultant, speaker, and/or researcher for Allergan, Coria, Galderma, Graceway, Intendis, Leo Pharma, Medicis, Onset Therapeutics, Ortho Dermatology, PharmaDerm, Promius, Quinnova, Ranbaxy, SkinMedica, Stiefel, Triax, Unilever, and Warner Chilcott.
Abstract
Dermatofibrosarcoma protuberans (DFSP) is an uncommon cutaneous neoplasm with intermediate- to low-grade malignancy, but can be a surgical challenge. DFSP is characterized by a storiform proliferation of bland-appearing cells that diffusely infiltrate the dermis. Due to these characteristics, it has a high potential for wide and deep extensions into the deep dermis. Although the majority of DFSPs are cured by Mohs micrographic surgery, tumor size and disfigurement may be a limiting factor for such a procedure. Moreover, even when surgery is utilized, there are cases where positive margins remain, in which case imatinib would be an option, often in combination with surgery. Imatinib can decrease tumor size preoperatively and help to improve postsurgical aesthetic appearance and minimize functional impairment. This article is a review of literature examining the indications for the use of imatinib in DFSP lesions and understanding the pathophysiology behind the treatment.
Introduction
Dermatofibrosarcoma protuberans (DFSP) is relatively uncommon spindle cell sarcoma of fibroblastic/myofibroblastic differentiation, accounting for 2 to 6 percent of all soft tissue sarcomas, yet is the most frequently encountered sarcoma of skin.[1] DFSP affects both genders equally and most commonly arises in adulthood, with many cases observed among those 20 to 40 years of age.[1]
Importantly, pediatric cases have been noted with more than 160 cases of acquired childhood DFSP, and more than 35 cases of the congenital DFSP tabulated in recent literature reviews.[1–3] The trunk is the most common anatomical site affected by DFSP (42–72%); however, proximal extremities (16–30%) and head and neck (10–16%) are not uncommon sites.[1]
Classically, DFSP presents as an asymptomatic, pink, red, yellow, or skin-colored plaque that is very indurated on palpation, and often develops isolated or multilobular surface nodularity later on as growth of the tumor progresses. As the neoplasm is dermal in origin and overlying visible skin changes may be subtle, there is often a significant delay before treatment is sought, resulting in large tumors at the time of presentation to a clinician. Histologically, DFSP is characterized by a storiform proliferation of bland-appearing spindle cells, low mitotic activity, and irregular tentacle-like extensions that randomly and diffusely infiltrate deep dermis and beyond.[1] Due to its infiltrative growth pattern, DFSP exhibits a high potential for wide and deep extension from deep dermis into subcutaneous fat, and not uncommonly into fascia and muscle, and sometimes bone. Notably, the extent of histological extension is markedly beyond the confines of the visible and palpable lesion.[1] Although the potential for metastasis of the classic form of DFSP is low (0.5%), metastatic DFSP can occur, especially with fibrosarcomatous transformation (DFSP-FS) or after multiple local recurrences due to inadequate surgical excisions.[1,4] The lung appears to be the most common site of involvement with metastatic DFSP, although other sites, such as the brain, pelvis, and rarely lymph nodes, have been observed.[1,5]
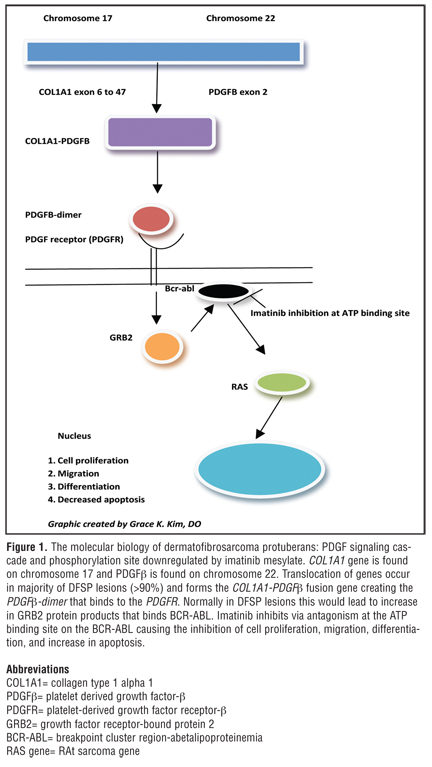
Surgical excision has been the most common therapeutic approach; however, local recurrences are common even with wide margins of excision, primarily due to the elusive and extensive infiltrative growth of DFSP.[1] Imatinib mesylate, an orally administered tyrosine kinase breakpoint cluster region-abetalipoproteinemia (BCR-ABL) inhibitor, which also affects ABL-related kinase, KIT, platelet-derived growth factor beta (PDGFbeta), and PDGF receptor-alpha, has been very effectively used to treat gastrointestinal (GI) stromal tumors (GISTs) and chronic myelogenous leukemia.[2,6] Coincidentally, the stimulation of expression of PDGF receptor beta (PDGFR beta) on the cell surface of many DFSP tumors has been noted, related to pathogenetic expression of the fusion gene collagen type 1 alpha 1 gene (COL1A1)-PDGF beta, which occurs secondary to specific cytogenetic abnormalities.[1,2,6] It has been reported that greater than 90 percent of DSFP tumors possess supernumerary ring chromosomes or a unique translocation involving chromosomes 17 and 22 t(17;22)(22;q13), which fuse the COL1A1 gene on chromosome 17 with the PDGF beta-chain gene on chromosome 22.1 PDGF beta, a tissue growth factor, encodes the beta-chain of PDGF, which is a ligand for tyrosine kinase PDGFR located on the cell surface. The aberrant fusion gene, COL1A1-PDGF beta, is under the control of the COL1A1 promoter, with the resulting fusion protein processed into mature PDGF beta in DFSP cells, leading to autocrine stimulation of PDGF beta R on the surface of DFSP cells and subsequent cellular proliferation and tumor growth.[1,2,6] As imatinib mesylate is an orally active selective tyrosine kinase inhibitor with activity against specific kinases, its ability to inhibit PDGFR protein-tyrosine kinase prompted interest in this drug for treatment of DFSP.[1]
This installment of Questions, Challenges, Controversies reviews available studies on the use of imatinib mesylate for DFSP, including metastatic DFSP and DFSP-FS, in both adults and children. Overall, imatinib mesylate has been used most commonly for locally advanced and metastatic DFSP, often as adjuvant therapy and sometimes as monotherapy.[1,2,6–8] Emphasis is placed on evaluating the range of therapeutic responses that have been reported and defining what might be done to predict when response to imatinib mesylate may or may not occur. Interestingly, when used to treat GISTs, imatinib mesylate produces a response rate of 49 to 83 percent (depending on the exact exon mutation type) in KIT-mutated GISTs and essentially no response in PDGF alpha-mutated GISTs, supporting the value of confirming mutation types as a means of predicting response prior to initiating therapy.[9] As the COL1A1-PDGF beta fusion product is detectable by reverse-transcriptase polymerase chain reaction (RT-PCR), pretreatment testing of DFSP tumors for specific cytogenetic abnormalities may also be of benefit in predicting response to therapy with imatinib mesylate.[1]
What are the conventional options used for treatment of DFSP and what is their success rate?
The primary treatment option for DFSP is surgical excision.[1] Most reports are of cases treated with standard surgical excision using a range of surgical margins, with some reports evaluating use of Mohs micrographic surgery for DFSP.[1,2] After standard surgical excision, local recurrence rates have been reported to be as high as 60 percent.[1] A tabulation of multiple studies evaluating surgical excision of DFSP depict a local recurrence rate of 39.7 percent in reports noting undefined or conservative surgical margins (N=116), 8.8 percent in reports describing wide surgical margins (N=661), and 1.5 percent in reports utilizing Mohs micrographic surgery (N=327), with the duration of follow up three years or greater in most of the cited reports.[1] The local recurrence rate of DFSP after wide surgical excision is 50 to 75 percent, likely due to the difficulty of resecting large anatomical areas due to functional and cosmetic considerations.[1] With standard surgical excision, use of wide surgical margins of 2 to 2.5cm to 3 to 3.5cm has been suggested to optimize the likelihood of complete tumor resection.[1] Mohs micrographic surgery appears to be a viable option, especially if performed by a dermatological surgeon experienced in using this technique for DFSP, including cases on the head and neck and in children.[2]
As DFSP is radiosensitive, radiation therapy has been used for primary or locally recurrent DFSP, usually as preoperative or postoperative adjuvant therapy.[1] This approach may be helpful in cases with narrow or positive surgical margins, or when foci of nonresectable tumors are present.[1] Overall, radiation therapy has been shown to improve local control rate as compared to surgery alone in selected cases; however, the total number of tabulated patients undergoing combined treatment with both surgery and radiation therapy is relatively low (N=86).[1] Nevertheless, radiation therapy appears to play an important role in selected clinical situations.
What are the histological subtypes of DFSP and is there clinical significance related to any of these subtypes?
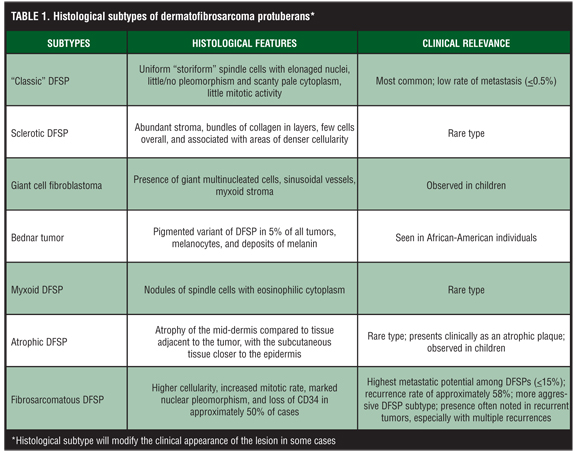
There is a variety of histological subtypes of DFSP that have been described in literature (Table 1). The different histological types are “classic” DFSP, sclerotic DFSP, giant cell fibroblastoma (GCF), Bednar tumor, myxoid DFSP, atrophic DFSP, and fibrosarcomatous DFSP (DFSP-FS). Approximately 90 percent of DFSP are considered to be tumors of low-grade malignancy called the “classic” form of DFSP,10 which is characterized by a growth pattern of uniform “storiform” spindle cells with elongated nuclei showing little to no pleomorphism and scanty pale cytoplasm with little to no mitotic activity. Sclerotic DFSP is characterized by an abundant stroma of collagen bundles forming layers, adopting a storiform pattern with few cells, and associated with areas of denser cellularity.[1] There is also GCF, a variant form of DFSP that primarily affects children, presenting histologically with multinucleated giant cells, sinusoidal vessels, and a myxoid stroma.[1] The Bednar tumor is a pigmented variant of DFSP occurring in five percent of all tumors seen in African-American patients with additional dispersal of melanin and melanocytes.[11] The presence of melanocytes may be secondary to “colonization” or may be possibly due to a neuroectodermal differentiation.[12] The myxoid type is a rare form of DFSP characterized by the presence of nodules of spindle cells with an eosinophilic cytoplasm. The atrophic type of DFSP is rare and presents clinically as an atrophic plaque, most often observed in children.[1,36] In this subtype, the mid-dermal thickness is atrophic compared to tissue adjacent to the tumor, and subcutaneous tissue below the body of this DFSP subtype is closer to the epidermis as compared to contiguous surrounding tissue.
{kind=link}
Rarely, DFSP can transform into the more aggressive DFSP-FS with a higher metastatic potential.[4,13] Approximately 3.2 to 15 percent of DFSPs contain the high-grade fibrosarcomatous component that typically occupies between 20 to 80 percent of the tumor mass.[10] Fibrosarcomatous changes in DFSP can lead to misdiagnosis as a fibrosarcoma, especially in partially sampled lesions. In the DFSP-FS type, the cells show higher cellularity, increased mitotic rate, and marked nuclear pleomorphism. There is also focal loss of CD34 observed in 50 percent of cases within the fibrosarcomatous regions, whereas the more banal classic-appearing storiform areas retain their staining pattern of CD34 positivity.[4] Focal necrosis, uncommon in the storiform component of DFSP, may also be present. Due to its more aggressive biological nature, DFSP-FS requires more extensive local treatment and closer follow up for possible development of metastases.
Most authors believe that the previously described variants of DFSP do not impart any prognostic significance, with the exception of the DFSP-FS type.[6] However, overall, all variants of DFSP have a metastatic rate of five percent, while the “classic” DFSP is reported to exhibit a 0.5-percent rate of metastasis.[14] It is not entirely clear whether this 4.5-percent difference is accounted for by differences in methods of data collection, reflects the impact primarily of fibrosarcomatous transformation, or is due to true differences among the various DFSP variants. Regardless, the sarcomatous histological changes seen in DFSP-FS are clinically relevant as they have been correlated with a worse prognosis as compared to “classic” DFSP.[4] Fibrosarcomatous variations within DFSP have been found in reviews to have a greater propensity for local recurrence (58%) and metastasis (15%).15 Interestingly, the risk of transformation into the fibrosarcomatous type is higher with recurrent DFSP.[15]
What are the specific cytogenetic abnormalities that have been described in many DFSP tumors? How do these findings correlate with potential mechanisms of action with imatinib mesylate in the treatment of DFSP?
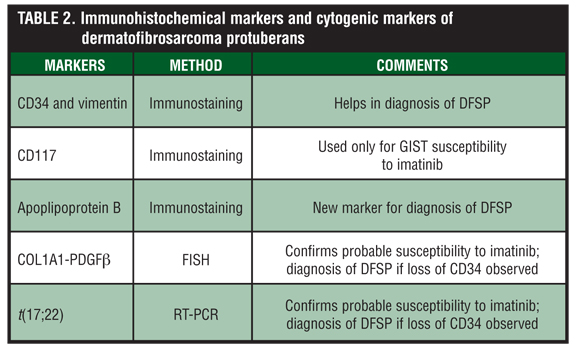
Cytogenetically, DFSP is characterized by a reciprocal translocation t(17;22)(q22;q13), or the supernumerary ring chromosomes derived from t(17;22). The consequence of this translocation is the fusion between the gene for COL1A1 on chromosome 17q and the gene encoding platelet-derived growth factor beta polypeptide (PDGF beta) on chromosome 22q (Figure 1).[1] PDGF beta is placed under the control of the COL1A1 promoter and the resulting fusion protein is processed into the mature PDGF beta in cells of DFSP leading to an autocrine stimulation of the PDGF receptor (PDGFR) on the cell surface. Therefore, this constant stimulation is responsible for the cyclic regeneration and proliferation of the tumor cells.[1] This molecular alteration is fundamental to the development of DFSP. Imatinib disrupts this pathway by inhibition of PDGF beta-PDGRT beta signaling pathway. The COL1A1-PDGF beta fusion product can be detectable by RT-PCR or fluorescent in-situ hybridization (FISH) to determine susceptibility to imatinib. The fibrosarcomatous variant (DFSP-FS), lacking the t(17;22) has been reported to exhibit decreased or absent response to imatinib.[16] In addition, approximately eight of DFSPs lack the fusion transcription, and other unidentified genes may be involved.[17] There have also been other studies demonstrating partial remission rates and inconsistent responses to imatinib in selected DFSP cases.[18,19] Reasons for these less-than-optimal therapeutic responses to imatinib that have sometimes been observed may be a resistance mutation in the PDGF beta receptor and possibly other signaling pathways of PDGFRs.[20] Reports have also detected COL1A1-PDGF beta fusion transcript present in the resected tumor tissue and may still be identified in histological remission.[2] The COL1A1-PDGF beta fusion product can also be utilized in the diagnosis of fibrosarcomatous transformation, metastatic DFSP, and atypical DFSP cases, where immunomarkers, such as CD34, have been lost.[21] The detection of COL1A1-PDGF beta fusion transcripts confirms the diagnosis of DFSP and helps direct the management of these cases (Table 2). In addition, some investigators have suggested using plasma PDGFB levels to monitor clinical response.[22]
{kind=link}
{kind=link}
What is imatinib mesylate and what does it do mechanistically to inhibit DFSP tumor growth?
Imatinib mesylate (imatinib) is an inhibitor of the BCR-ABL gene, an oncogene fusion protein associated with the Philadelphia chromosome (Ph+). This abnormality is seen with chronic myeloid leukemia (CML) and also with acute lymphoblastic leukemia (AML). Imatinib is first-line treatment of CML and metastatic or unresectable GIST, which also has aberrantly activated tyrosine kinases (ABL and KIT).[1] PDGF is overexpressed in DFSP cells because of COL1A1-PDGF beta oncogene formation. The PDGF beta-PDGRT beta signaling pathway in the development of DFSP is the target of therapy for imatinib. The ability of imatinib to inhibit the receptor tyrosine-kinase for PDGF (PDGFR), c-KIT (stem-cell factor), and ABL kinases renders it useful in the treatment of DFSP.[1] Imatinib inhibits the activity of the PDGF beta protein that causes increased proliferation by attaching to the ATP binding site needed for auto-phosphorylation of tyrosine kinases (Figure 1). This binding decreases the enzymatic activity in DFSP cells and inhibits their ability to divide and grow. Furthermore, the reduced enzymatic activity of imatinib has been shown to induce apoptosis in tumor cells.[23] Both in-vitro and in-vivo data have shown that blocking of the PDGF beta autocrine loop in DFSP with imatinib leads to reversible reduction of DFSP cell line growth.[23,24] This indicates that the activation of the PDGF beta R tyrosine kinase is essential to the pathogenesis of the disease and vital for tumor cell growth. Investigators have observed DFSP lesions with an increase in hypocellular and acellular areas post-imatinib therapy, leading many to believe in the apoptosis theory as the mechanism responsible for decreasing tumor size.[25] Other theories postulate that imatinib may change the phenotype of DFSP cells reducing proliferation and tumor size, thus making the lesion more amenable to complete surgical resection.[25]
What are the immunohistochemical findings in DFSP? Are any of these immunohistochemical findings of clinical significance? Do any immunohistochemical findings correlate with response or lack of response of DFSP to imatinib mesylate?
Approximately 80 to 90 percent of DFSP express CD34, which is the most useful immunohistochemical marker in the differential diagnosis.[25] Immunohistochemically, DFSP generally stains positive for CD34 and vimentin but negative for factor XIIIa (Table 2). In addition, apolipoprotein B has been proposed as a new marker for DFSP.[26] CD34 is expressed in hematopoetic progenitors including leukemias and in endothelial cells in benign and malignant tumors.[15] CD34 helps differentiate DFSP from dermatofibroma and other soft tissue tumors. It is also expressed in other sarcomas, such as inflammatory myofibroblastic tumor, myofibrosarcoma, epithelioid sarcoma, angiosarcoma, and even in some benign fibrohistiocytic lesions.[25] Approximately 10 to 20 percent of DFSPs are negative for CD34, with higher incidences in the DFSP-FS variant.[25] Immunomarkers do not seem to be an indicator for response to imatinib, but are of great importance to the pathologist in differentiating tumors that are part of the differential diagnosis.
Although immunomarkers, such as CD117, are useful markers in GI tumors, more studies with DFSP immunohistochemical markers correlating response to imatinib are needed. Currently, imatinib is approved for the treatment of patients with KIT (CD117)+ unresectable and/or metastatic malignant GIST.[27] The response to imatinib of GISTs with mutations in the KIT receptor and the PDGFA receptor are 80 and 7 percent, respectively.[28] Those with KIT mutations stain strongly with CD117 while those with PDGF alpha mutations tend to be CD117 negative. In addition, imatinib results in a response rate of up to 83 percent in KIT-mutated GISTS (83% for mutations in the exon 11 and 49% for mutations in exon 9), whereas no response is observed in the PDGF alpha-mutated GISTS. One study examined 37 cases of DFSPs and 13 cellular DFs and found that all cases were negative for CD117.[9] These authors suggest that CD117 should not be used as an immunohistochemical marker for the eligibility of imatinib therapy.
In what clinical situations has imatinib mesylate been reported or studied for use in DFSP? What is the range and nature of therapeutic responses reported with this therapy for DFSP?
Although the majority of DFSPs are cured by Mohs micrographic surgery, tumor size and disfigurement may be a limiting factor for such a procedure. Moreover, even when surgery is utilized, there are cases where positive margins remain, in which case imatinib would be an option, often in combination with surgery. Additionally, long-term control is a challenge for DFSP with a high tendency for local recurrence despite the use of radiotherapy to improve outcome. Imatinib can decrease tumor size preoperatively and help to improve postsurgical aesthetic appearance and minimize functional impairment.[7] In 2006, the United States Food and Drug Administration approved imatinib for unresectable, recurrent, and/or metastatic DFSP in adults. In order to maximize the opportunity for beneficial results, evaluation of t(17;22) translocation should be conducted by a multidisciplinary team for patients to be considered for imatinib therapy. Besides as a treatment in metastatic disease, a high percentage of advanced DFSP cases (fibrosarcomatous subtypes) have also been reported to respond to imatinib therapy.[7] Side effects are mild and well tolerated. However, there are still unanswered questions regarding the optimal dosage and duration of imatinib therapy, with a dosage range of 400 to 800mg/day reported. Some studies have shown that 400mg once daily is sufficient enough for therapeutic response.
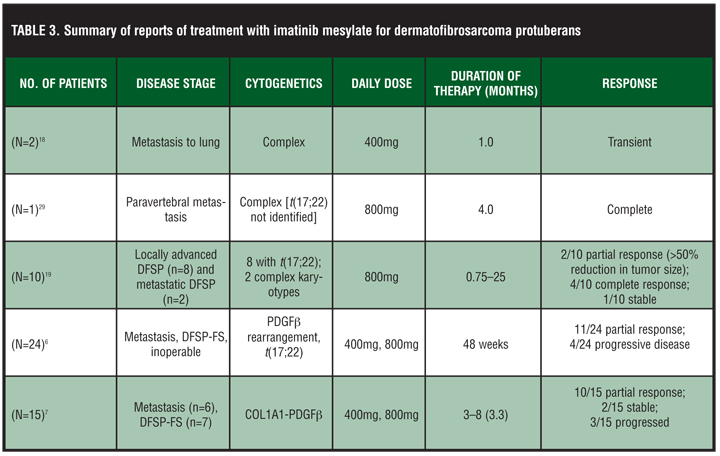
There have been many case reports of imatinib in the treatment of metastatic, DFSP-FS, and locally advanced DFSP (Table 3). The first case report of treatment and response to imatinib described two patients with metastatic and inoperable DFSP using 400mg daily with only one patient exhibiting t(17;22).18 In the patient having the t(17;22), resolution of numerous small lung nodules and a 9.7cm paratracheal mass was observed. At the time of the report, this patient experienced continued improvement in his residual disease after an additional two months of imatinib therapy.[18] The patient with the translocation experienced an impressive partial response for six months. Another case report describing a patient with metastatic DFSP revealed 75-percent tumor shrinkage after four months of treatment with imatinib 400mg twice daily.[29] Response to therapy was assessed by fluorodeoxyglucose-positron emission tomography (FDG-PET) scanning and magnetic resonance imaging (MRI). The hypermetabolic uptake of FDG fell to background levels within two weeks of treatment, and the tumor volume decreased by more than 75 percent over four months of therapy, allowing resection of the metastatic mass.29 The resected specimen showed no residual tumor, indicating a complete histological response.[29] Most case reports have ranged anywhere from one to 51 months (average 8–10 months) with imatinib doses of 400 or 800mg/day. When case reports have been pooled together, the usual time to initial response is approximately three months, but responses as early as one month have been noted in some cases.30 Before therapy, the majority of case reports explore cytogenetic abnormalities involving COL1A1-PDGF beta by FISH or t(17;22) by RT-PCR to confirm susceptibility to imatinib.
{kind=link}
The first large-scale clinical experience with imatinib in the treatment of DFSP was described in a multicenter Phase II trial evaluating use in advanced neoplasms expressing imatinib-sensitive protein tyrosine kinases.[19] This trial included 12 patients with advanced DFSP treated with imatinib 400mg twice daily, demonstrating a clinical benefit of 83.3 percent (50% partial response rate, 33.3 percent complete remission rate).[19] Two other Phase II trials of imatinib (400 to 800mg/day), one in Europe by the European Organization for Research and Treatment of Cancer (EORTC) and one in North America by the Southwest Oncology Group (SWOG), examined local or metastatic DFSP with 14-week progression-free rate as the endpoint.[6] There were a total of 24 patients (17 EORTC, 8 SWOG). Eleven patients (46%) had partial response and four had progressive disease. The one-year overall survival (OS) rate was 87.5 percent.[6] Response rates reached 50 percent with no difference between 400 versus 800mg/day at Day 6. This study reported the first ever data regarding progression-free rate and OS.
In another study, 15 patients with locally advanced/initially inoperable and/or metastatic DFSP were treated with imatinib 400 to 800mg daily averaging 3.3 months of treatment before surgical excision.[7] The median follow up time was 16 months (range 4–18 months). The presence of molecular target was confirmed in every case prior to the onset of therapy.[7] The two-year progression-free survival rate was 60 percent, and the one-year OS rate was 78 percent.[7] The data from this study suggested that 400mg/day of imatinib may be just as efficacious in treating DFSP as the higher dose, which was also noted in the European and United States trials. This study also showed that a high percentage (71%) of higher grade DFSP-FS, which are more difficult to treat with conventional methods and characteristically exhibit a worse prognosis, were responsive to imatinib therapy.[7]
What information is available on the use of imatinib mesylate in children?
Currently, imatinib is not indicated for pediatric cases (<18 years) of DFSP.[2,31] However, pediatric CML studies with Phase I/II data are available to appropriately dose the drug in children.[31] The safety and efficacy of imatinib has been demonstrated only in children with newly diagnosed, recurrent, or resistant Ph+ chronic phase CML.[31] Pediatric cases of DFSP have been reported with the congenital type and acquired type.2 The cause of congenital DFSP is unknown. Trauma has been implicated in the pathogenesis of up to 20 percent of cases in childhood DFSP.[32] It has also been associated with enlargement or evolution of congenital DFSP in 10 percent of cases.[33,34] Pediatric cases of DFSP are frequently misdiagnosed and mistaken for a vascular malformation or tumor. The aberrant t(17;22) has been reported positive in approximately 69 percent of congenital DFSP.[33,35,36] Clinical improvement has been seen in an individual as young as 18 months treated with imatinib for unresectable disease with a partial response.[35] The patient was treated initially with imatinib 400mg/m2/day, and later titrated upward to 520mg/m2/day. Major clinical improvement was evident within four weeks of treatment.
Follow-up MRI after 23 weeks of therapy demonstrated reduction in the subcutaneous thickness of the tumor. Imatinib can also be used as adjuvant therapy to prevent relapses in children, but it has not been shown definitively to be effective.[2] However, case reports have reported imatinib to improve relapse-free survival rates in pediatric patients post-surgery.[2] Molecular screening should be used as a guide for imatinib therapy post-surgery in order to assure continuous remission.[2] Side effects of imatinib in pediatric patients have been noted to be mild. The overall safety profile in 93 pediatric patients studied was similar to that in adult patients.[31] In contrast, musculoskeletal pain occurred less frequently in pediatric patients than in adults, and peripheral edema was not reported in pediatric patients.[31] Most frequently reported adverse effects include nausea and vomiting.[31] Administration difficulties due to bitter taste have been reported and rectal administration of a double dose of imatinib mesylate may be an alternative.
What are the major risks and adverse reactions associated with the use of imatinib mesylate therapy?
Many clinical studies have reported mild but tolerable side effects with imatinib. The most common adverse events noted were fluid retention/edema, anemia, fatigue, nausea, skin toxicity, thrombocytopenia, vomiting, neutropenia, and diarrhea.7 Management of edema may include diuretics or reduction of imatinib dosage. If severe fluid retention develops, imatinib therapy should be interrupted until complete resolution occurs.[31] If substantial increases in bilirubin (>3 times upper limit normal [ULN]) or hepatic transaminase concentrations (>5 times ULN) occur, imatinib should be discontinued until bilirubin or transaminase concentrations decrease to <1.5 or <2.5 times ULN, respectively.[31]
Imatinib can also induce major cutaneous side effects, such as bullous skin reactions, and it is recommended to discontinue imatinib if this is observed. Re-initiation of imatinib therapy can be tolerated in some patients at a lower daily dosage (with or without corticosteroids or antihistamines) following resolution or lessening of the bullous skin reaction.[31] Adults previously receiving a dosage of 400, 600, or 800mg daily may resume therapy at a dosage of 300, 400, or 600mg daily, respectively.[31] Pediatric patients previously receiving a dosage of 260 or 340mg/m2/day may resume therapy at a dosage of 200 or 260mg/m2/day, respectively.[31]
What are the major “take-home points” that summarize for the clinician newer information on DFSP and the role(s) of imatinib mesylate in the treatment of DFSP?
DFSP is a rare cutaneous sarcoma, usually of low-grade biologic behavior, that often exhibits dysregulated PDGF production derived from t(17;22). This translocation resulting in a fusion product of COL1A1-PDGF beta is the target of imatinib therapy. Therefore, the presence of this translocation fusion product is best ascertained before initiation of treatment with imatinib. Surgical resection can be confounded depending on the location and size of the tumor, often leading to disfigurement, morbidity, and loss of function. Additionally, tumor-positive margins are not uncommon, even with wide excision. Although treatment with Mohs micrographic surgery offers high clearance rates, imatinib may be used to decrease tumor size and maximize surgical results. Imatinib may also allow for complete resection in inoperable cases and is an option for metastatic disease. Overall, imatinib exhibits a favorable side effect profile, with many adverse effects circumvented by temporary discontinuation of therapy and dosage reduction. However, there are still unanswered questions concerning optimal dosage, treatment duration, and determination of clinical endpoint. Prompt diagnosis and treatment are essential to maximize functional and aesthetic outcomes. Ultimately, patients with a history of DFSP undoubtedly must be carefully monitored for recurrence, with a multidisciplinary approach to evaluation and treatment recommended.
References
1. Lemm D, Mugge LO, Mentzel T, et al. Current treatment options in dermatofibrosarcoma protuberans. J Cancer Res Clin Oncol. 2009;135:653–665.
2. Gooskens SLM, Oranje AP, van Adrichem LNA, et al. Imatinib mesylate for children with dermatofibrosarcoma protuberans. Pediatr Blood Cancer. 2010;55:369–373.
3. Love WE, Keiler SA, Tamburro JE, et al. Surgical management of congenital dermatofibrosarcoma protuberans. J Am Acad Dermatol. 2009;61:1014–1023.
4. Mentzel T, Behm A, Katenkamp D, et al. Fibrosarcomatous (“high grade”) dermatofibrosarcoma protuberans: clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am J Surg Pathol. 1998;22:576–587.
5. Rutgers EJ, Kroon BB, Albus-Lutter CE, et al. Dermatofibrosarcoma protuberans: treatment and prognosis. Eur J Surg Oncol. 1992;18:241–248.
6. Rutkowski P, Van Glabbeke M, Rankin CJ, et al. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol. 2010;28:1772–1779.
7. Rutkowski P, Debiec-Rychter M, Nowecki ZI, et al. Treatment of advanced dermatofibrosarcoma protuberans with imatinib mesylate with or without surgical resection. JEADV. 2010:1–7.
8. Lemm D, Mugge LO, Hoeffken K, et al. Remission with imatinib mesylate treatment in a patient with initially unresectable dermatofibrosarcoma protuberans—a case report. Oral Maxollofac Surg. 2008;12:209–213.
9. Labonte S, Hanna W, Bandarchi-Chamkhaleh B. A study of CD117 expression in dermatofibrosarcoma protuberans and cellular dermatofibroma. J Cutan Pathol. 2007;34:857–860.
10. Bowne WB, Antonescu CR, Leung DHY et al. Deramtofibrosarcoma protuberans. A clinicopathologic analysis of patients treated and followed at a single institution. Cancer. 2000;88:2711–2720.
11. Bednar B. Dermatofibrosarcoma protuberans of the head and neck. Arch Otolaryngol. 1957;111:132–138.
12. Goncharuck V, Mulvaney M, Carlson J, et al. Bednar tumor associated with dermal melanocytosis: melanocytic colonization or neuroectodermal multidirectional differentiation? J Cut Pathol. 2003;30:147–151.
13. Wrotnowski U, Cooper PH, Shmookler BJ. Fibrosarcomatous change in dermatofibrosarcoma protuberans. Am J Surg Pathol. 1998;12:287–293.
14. Brenner W, Schaefler K, Chabra H, Postel A. Dermatofibrosarcoma protuberans metastatic to a regional lymph node: report of a case and review. Cancer. 1975;36:1897–1902.
15. Stefanos V, Razis L, Razis E. Imatinib in the treatment of dermatofibrosarcoma protuberans. Biologics. 2007;1(4):347–353.
16. Handolias D, McArthur G. Imatinib as effective therapy for dermatofibrosarcoma protuberans: proof of concept of the autocrine hypothesis for cancer. Future Oncology. 2008;4(2):211–217.
17. Sirvent N, Maire G, Pedeutour F. Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer. 2003;37:1–19.
18. Maki RG, Awan RA, Dixon RH, et al. Differential sensitivity to imatinib of 2 patients with metastatic sarcoma arising from dermatofibrosarcoma protuberans. Int J Cancer. 2002;100:623–626.
19. McArthur GA, Demetri GD, van Oosterom A, et al. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: imatinib target exploration consortium study