Jason J. Emer, MD; Shayna Solomon, MD; Stephen E. Mercer, MD, PhD
Drs. Emer and Mercer are from Department of Dermatology, Mount Sinai School of Medicine, New York, New York; Dr. Solomon is from SUNY Downstate, University Hospital of Brooklyn at Long Island College Hospital, Brooklyn, New York
Disclosure: The authors report no relevant conflicts of interest.
Abstract
Multiple cutaneous and uterine leiomyomatosis, also known as Reed’s syndrome, is an autosomal dominant genetic condition. Affected individuals have an increased predisposition to develop benign smooth muscle tumors (leiomyomas) in the skin and uterus. Affected females frequently develop uterine leiomyomas (fibroids) that are larger and more numerous and emerge earlier than those in the general population. Subsets of these patients are at risk for renal cell cancer and have been determined to have mutations in the fumarate hydratase gene. In individuals or families without renal cell cancer, the syndrome may be referred to as multiple cutaneous leiomyomatosis or multiple cutaneous and uterine leiomyomatosis. The term hereditary leiomyomatosis and renal cell cancer refers to families with an increased prevalence of smooth muscle tumors and renal cell cancer as a result of the fumarate hydratase genetic defect. In this article, the authors introduce a case of a young woman who presented with multiple, intermittently painful, cutaneous leiomyomas and a history of large uterine fibroids previously causing anemia and requiring surgical intervention. Further investigation revealed a family history of mutations in the fumarate hydratase gene. The patient is currently being monitored by the National Institutes of Health.
(J Clin Aesthet Dermatol. 2011;4(12):37–42.)
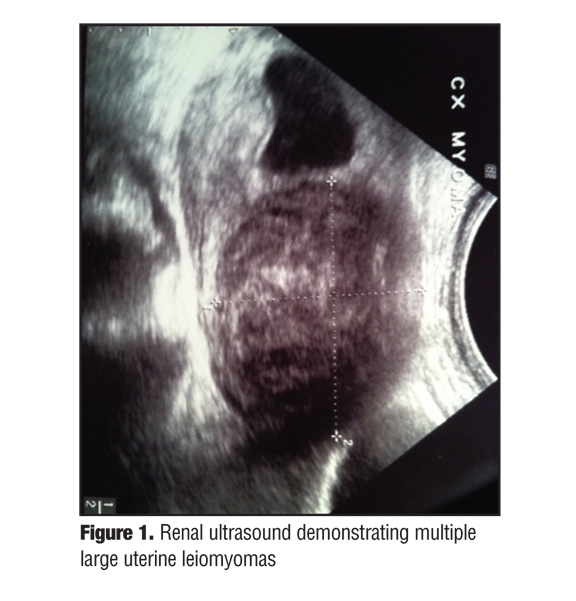
A 24-year-old Hispanic woman presented to the authors’ outpatient dermatology clinic complaining of a multiple-year history of intermittently painful bumps on her mid-back and left shoulder. Recently, the lesions had become more painful to touch and increased in both size and number. The patient’s past medical history was remarkable for anemia that required treatment with blood transfusions. She also had a history of multiple myomectomies for uterine fibroids, starting as early as 21 years old (Figure 1). A family history revealed that her father, maternal uncle, and sister also had a history of multiple painful “skin bumps.”
{kind=link}
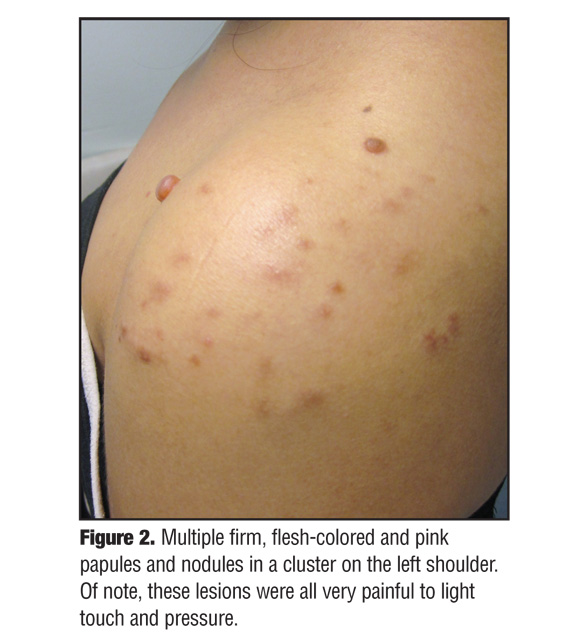
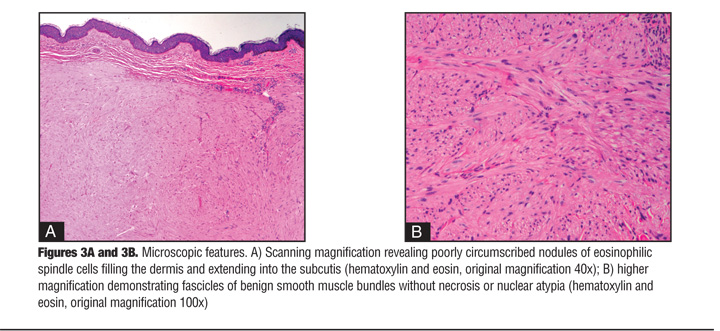
Physical examination revealed multiple flesh-colored to pink smooth, nonmobile papules and nodules arranged in a cluster on the left shoulder (Figure 2). The papules and nodules were firm and tender and ranged in size from 0.5 to 2cm in diameter. The most prominent and painful nodules were excised and sent for histological analysis. Microscopic examination revealed dense dermal nodules composed of eosinophilic spindle cells (Figure 3a). Closer examination exposed elongated cells with abundant eosinophilic cytoplasm arranged in fascicles (Figure 3b). The blunt-ended, cigar-shaped nuclei were uniform with only rare normal mitoses. The findings were consistent with a diagnosis of benign leiomyomas.
{kind=link}
{kind=link}
Given the patient’s unique clinical presentation in conjunction with her surgical and family histories, the authors suspected a diagnosis of Reed’s syndrome. The patient was referred for genetic counseling and for evaluation of renal malignancy. Renal ultrasound and computed tomography (CT) were negative for renal pathology. At the present time, the patient, along with her extended family, is being evaluated by the National Institutes of Health (NIH) for genetic defects in the fumarate hydratase (FH) gene.
Introduction
The coexistence of benign smooth muscle growths in the skin and in the uterus is known as Reed’s syndrome.[1] In 1973, Reed et al[2] reported on two families in which members of successive generations demonstrated cutaneous leiomyomas, uterine leiomyomas, and/or leiomyosarcomas, establishing an autosomal dominant pattern of inheritance.[2,3] Many acronyms or related terms have been cited in the literature for this syndrome or closely related entities, such as multiple cutaneous leiomyomatosis (MCL), multiple cutaneous and uterine leiomyomatosis (MCUL), and/or leiomyomatosis cutis et uteri. Further, others have reported the association of MCUL with papillary type 2 renal cell cancer (RCC).[4] The disease predisposing gene has been identified as FH, a gene encoding an enzyme that operates in the mitochondrial citric acid cycle (Krebs cycle) and is intimately involved in cellular energy metabolism.[5,6]
There are three known tumor types originating from smooth muscle cells. The tumors in Reed’s syndrome appear to be of the common type originating from the arrector pili muscles surrounding hair follicles.[7,8] Since cutaneous leiomyomas are not exceptionally common, their presence—whether single or in multiplicity—should raise suspicion of underlying uterine leiomyomas and the possibility of an increased risk of RCC.[9] Increased clinical awareness is important due to the association between cutaneous disease, uterine fibroids, and the propensity toward aggressive RCC in certain populations.
Etiology
Familial studies have localized the predisposition for Reed’s syndrome to chromosome 1q42.3–43, namely the MCUL 1 locus of the gene encoding FH.[10] The discovery occurred shortly after researchers reported that some individuals with MCL were also at risk of developing an aggressive form of RCC.[11] In 2001, researchers localized the hereditary leiomyomatosis and renal cell cancer (HLRCC) locus to a region on chromosome 1q42–44.[12] Subsequently, more precise analysis narrowed down the location to a 14-cM region on 1q42.3–43.[5,12,13] From this, researchers were able to identify germline and somatic mutations along with loss of heterozygosity in FH of tumor tissue.[6] Subsequently, the Multiple Leiomyoma Consortium proposed that loss of function of the FH protein is the basis of tumor formation in HLRCC and named the gene as the susceptibility focus for HLRCC.[12]
Mutations of the missense, nonsense, frameshift, insertion, and splice-site types have been discovered in the FH gene.[6] Mutation analysis of 21 North American families with HLRCC identified germline mutations of the FH gene in 100 percent (21/21).[14] Of these germline mutations, the majority (71.4%; 10/14) were missense with the remaining being insertion (7.1%; 1/14), nonsense (14.3%; 2/14), and splice-site (7.1%; 1/14) mutations, all located along the entire length of the FH gene coding region. Of families with HLRCC, 62 percent (13/21) had RCC and 76 percent (16/21) had cutaneous leiomyomas. Of female FH mutation carriers, 100 percent (22/22) had uterine fibroids. This study demonstrated the variability in the cutaneous manifestations of HLRCC. In total, these researchers have identified 31 different germline FH mutations in 56 families with HLRCC, with a detection rate of 93 percent (52/56).
Other reports have identified FH mutations in approximately 75 percent of MCUL cases.[15] In one case series, evidence of germline FH mutations was found in 89 percent of women with multiple skin leiomyomas.[16] Of those with FH mutations, 69 percent had both skin and uterine leiomyomas, 15 percent had only skin leiomyomas, seven percent had only uterine leiomyomas, and nine percent were clinically unaffected. In this series, 100 percent of male patients had skin leiomyomas.
FH alterations are believed to correlate with tumor formation in families with HLRCC; however, the pathological mechanism for this relationship is not entirely clear. A cell that lacks functional FH has a subsequent metabolic derangement due to its defective Kreb cycle.[17] It has been suggested that pseudohypoxia due to defective enzymatic metabolism may drive cellular transformation and tumorigenesis.[18] Additionally, recent genetic analysis suggests that FH may act as a tumor suppressor gene, but the consequence thereof has yet to be determined.[15]
Clinical Presentation
The clinical characteristics of HLRCC may include cutaneous leiomyomas, uterine leiomyomas, and/or renal tumors. As described above, there is variability in the cutaneous presentation. Reed’s syndrome classically manifests with solitary or multiple cutaneous leiomyomas, which appear as firm skin-colored or pink-brown papules or nodules up to 2cm in diameter and are often associated with pain.[9] Leiomyomas are benign tumors composed of smooth muscle fibers that arise from either the arrector pili of hair follicles, dartos muscle of genital skin, or smooth muscle of the vasculature.[19] A pseudo-Darier sign may be present, which is a transient piloerection or elevation of a lesion induced by rubbing. The clinical presentation of multiple lesions has been described in various patterns or distributions, such as bilateral and symmetric, clustered, linear, zosteriform, dermatomal-like, or disseminated.[19,20] Although commonly distributed over the trunk, the face may also be affected. It is important to consider a leiomyoma in any indolent nodular or papular growth associated with pain. An acute increase in size and/or number of lesions and/or an increase in pain should be suggestive of worsening disease or the rare possibility of malignant degeneration. The majority of leiomyomas will not spontaneously regress. Rather, most require surgical treatment for complete removal if the lesion is painful and/or cosmetically displeasing.
Diagnosis
The clinical diagnosis of cutaneous leiomyomas may be challenging and ultimately requires histopathological analysis. Although guidelines for screening do not exist, it may be difficult to differentiate a case of simple benign cutaneous leiomyomas from that of HLRCC. A high index of suspicion is required in order to ensure that patients are properly screened and receive appropriate diagnostic exams. MCL should raise suspicion for underlying disease and at least one cutaneous lesion should be histologically confirmed. On microscopic examination, smooth muscle fiber bundles composed of eosinophilic cytoplasm with elongated blunt-ended nuclei with little or no waviness (described as cigar- or eel-shaped) are interspersed with collagen within the dermis.[21] On cross-section analysis, perinuclear vacuolization may be visualized, but by electron microscopy, the smooth muscle cells will appear normal.[22] Special stains can be used to distinguish smooth muscle from collagen, since both will appear pink-red on hematoxylin-eosin stain. Masson’s trichrome will reveal smooth muscle as red and collagen as blue-green. Van Gieson stain highlights smooth muscle as yellow and collagen as red. Phosphotungstic acid–hematoxylin (PTAH) stain will be purple owing to presence of myofibrils in the specimen.[23] The markers of smooth muscle differentiation (desmin and actin) will be positive.[24]
Immunohistochemical staining will be negative for estrogen and progesterone receptors in cutaneous leiomyomas, but are positive in uterine leiomyomas.[25] FH gene enzyme activity in cultured skin fibroblasts or lymphoblastoid cells may demonstrate decreased activity (?60%).[26] As described previously, molecular genetic testing with sequencing analysis will demonstrate mutations in the FH gene in 80 to 100 percent of affected individuals.[16] If sequence analysis fails to demonstrate a mutation and disease is present, multiplex ligation-dependent probe amplification (MLPA) can be helpful to identify whole-gene deletion.[27] MLPA is a novel technique that can accurately detect gross gene deletions or duplications of DNA sequences. DNA samples can be rapidly screened for exon deletions and/or duplications of the FH gene by this method, providing a novel method to screen individuals as well as family members.[28]
Differential Diagnosis
The diagnosis of Reed’s syndrome can be difficult, given its varied cutaneous presentations. Nevertheless, accurate diagnosis is critical due to the potential for concealed malignancy. As leiomyomas are classically painful, the complete list of painful papulonodules should all be taken into consideration on initial evaluation.[29] The popular pneumonic “LEND AN EGG” represents a list of painful tumors of the skin that includes: leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomus tumor, and granular cell tumor.[30] Segmental distribution may suggest herpes zoster, which is often painful, but presents with an acute onset of vesicles on an erythematous base that develop into crusts.[31] Smooth muscle hamartomas classically appear as flesh-colored plaques—not papules or nodules—and will most often be present at birth and not uncommonly have associated hypertrichosis and hyperpigmentation.[32] Although not generally painful, other tumors, such as trichoepithelioma, lipoma, cylindroma, and poroma, must also be included in the differential given their similar presentations. However, each has a unique distribution and appearance.[33]
Treatment and Management
The treatment of cutaneous leiomyomas is dictated by the number of lesions and the degree of discomfort or cosmetic nuisance. Camouflage cosmetics (makeup) and avoidance of painful triggers, such as cold and/or pressure, may be all that is needed. When only a few lesions are present, surgical excision is the gold standard for complete removal, but may have a high rate of recurrence and possibly require skin grafting for larger lesions.[19] Recurrences have been reported to occur from six weeks to more than 15 years following excision.[22] Destructive methods, such as electrodessication or cryotherapy, may be employed for small, individual, or few lesions, but little benefit over excision has been demonstrated and unwanted scarring and recurrence may occur as a consequence.[34]
Medical management plays a limited role in hastening the formation of new lesions and facilitating the resolution of current ones, but can be utilized for symptomatic pain relief. Medications known to affect smooth muscle contraction, such as nitroglycerine, nifedipine, phenoxybenzamine, and doxazosin, can provide effective pain relief.[19] The theory behind this method of treatment is that muscle contraction and subsequent nerve stimulation is responsible for the characteristic stabbing, shocking, and/or striking pain described by a majority of patients found to have these tumors.[35–37]
Medications that target the activity of nerves are of special interest because of an increased nerve density within and around leiomyomas.[38] Gabapentin and topical analgesics, such as lidocaine or capsaicin, may be successful in patients with temperature-induced tenderness.[39]
Recent studies show promising results from novel therapies such as botulinum toxin injection and carbon dioxide laser ablation for pain control.[40–42] Botulinum toxin type A is used in various pain syndromes as it is believed to work by inhibiting the release of neuropeptides, including substance P and glutamate, thus reducing central pain signals.[43,44] Carbon dioxide laser ablation provides an alternative to invasive surgical techniques, facilitates myolysis, and offers pain relief.[42,45]
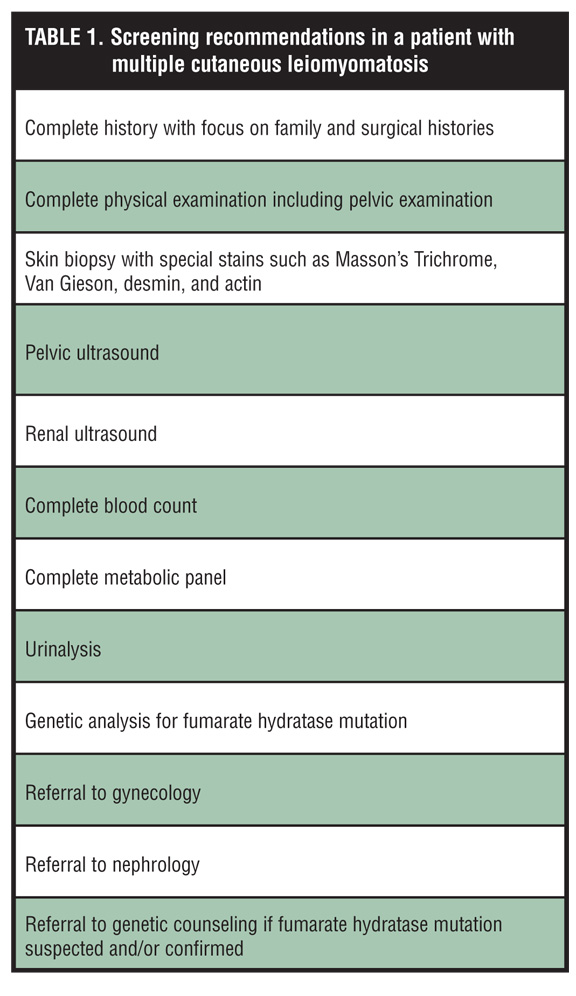
In addition, any individual presenting with cutaneous leiomyomas should be given a complete physical examination with a comprehensive history, including a focus on the family and surgical histories. All female patients with MCL should be evaluated for uterine disease. Currently, the NIH recommends evaluating all patients with leiomyomatosis for the presence of an occult renal malignancy.[26,46] Table 1 lists the authors’ screening recommendations for a patient with multiple cutaneous leiomyomas.
{kind=link}
Conclusion
Cutaneous leiomyomas are benign, smooth muscle tumors that may be a sign of underlying systemic disease. Increased awareness of the connection between cutaneous lesions and RCC can lead to life-saving early detection and treatment of an RCC that otherwise might have been allowed to develop undetected. It is critical that dermatologists perform a comprehensive evaluation of each patient presenting with cutaneous disease. Treatments for cutaneous lesions are only necessary if they exist in multiplicity, are enlarging or symptomatic, and/or are cosmetically displeasing. Appropriate surveillance with diagnostic testing for uterine and renal disease is warranted in rare cases of multiple, biopsy-proven cutaneous lesions. Genetic analysis of the FH gene is available through the NIH and should be performed in all cases of suspected or confirmed disease. Genetic counseling is also recommended for other members of the patient’s family. Publicizing reports of patients with HLRCC is essential to increase awareness of the correlation between cutaneous lesions and RCC, the ultimate goal of which is to promote early detection of underlying malignancy.
References
1. Hereditary leiomyomatosis and renal cell cancer. http://ghr.nlm.nih.gov/condition=hereditaryleiomyomatosisandrenalcellcancer. Accessed on April 17, 2011.
2. Reed WB, Walker R, Horowitz R. Cutaneous leiomyomata with uterine leiomyomata. Acta Derm Venereol. 1973;53:409–416.
3. Kloepfer HW, Krafchuk J, Derbes V, Burks J. Hereditary multiple leiomyoma of the skin. Am J Hum Genet. 1958;10:48–52.
4. Kiuru M, Launonen V. Hereditary leiomyomatosis and renal cell cancer (HLRCC). Curr Mol Med. 2004;4:869–875.
5. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95–106.
6. Alam NA, Olpin S, Leigh IM. Fumarate hydratase mutations and predisposition to cutaneous leiomyomas, uterine leiomyomas and renal cancer. Br J Dermatol. 2005;153:11–17.
7. Orellana Diaz O, Hernandez Perez E. Leiomyoma cutis and leiomyosarcoma: a 10-year study and a short review. J Dermatol Surg Oncol. 1983;9:283–287.
8. Badeloe S, Frank J. Clinical and molecular genetic aspects of hereditary multiple cutaneous leiomyomatosis. Eur J Dermatol. 2009;19:545–551.
9. García Muret MP, Pujol RM, Alomar A, et al. Familial leiomyomatosis cutis et uteri (Reed’s syndrome). Arch Dermatol Res. 1988;280:S29–S32.
10. Alam NA, Bevan S, Churchman M, et al. Localization of a gene (MCUL1) for multiple cutaneous leiomyomata and uterine fibroids to chromosome 1q42.3–q43. Am J Hum Genet. 2001;68:1264–1269.
11. Merino MJ, Torres-Cabala C, Pinto P, Linehan WM. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–1585.
12. Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410.
13. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825–829.
14. Wei MH, Toure O, Glenn GM, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet. 2006;43:18–27.
15. Alam NA, Rowan AJ, Wortham NC, et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet. 2003;12: 1241–1252.
16. Alam NA, Barcaly E, Rowman AJ, et al. Clinical features of multiple cutaneous and uterine leiomyomatosis: an underdiagnosed syndrome. Arch of Derm. 2005;141:199–206.
17. Badeloe S, van Geel M, van Steensel MA, et al. Diffuse and segmental variants of cutaneous leiomyomatosis: novel mutations in the fumarate hydratase gene and review of the literature. Exp Dermatol. 2006;15:735–741.
18. Sudarshan S, Pinto PA, Neckers L, Linehan WM. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104–110.
19. Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477–490.
20. Smith CG, Glaser DA, Leonardi C. Zosteriform multiple leiomyomas. J Am Acad Dermatol. 1998;38:272–273.
21. Kilpatrick SE, Mentzel T, Fletcher CD. Leiomyoma of deep soft tissue. Clinicopathologic analysis of a series. Am J Surg Pathol. 1994;18:576–582.
22. Fisher WC, Helwig EB. Leiomyomas of the skin. Arch Dermatol. 1963;88:510–520.
23. Spencer JM, Amonette RA. Tumors with smooth muscle differentiation. Dermatol Surg. 1996;22:761–768.
24. Miettinen M, Lehto VP, Virtanen I. Antibodies to intermediate filament proteins. The differential diagnosis of cutaneous tumors. Arch Dermatol. 1985;121:736–741.
25. McGinley KM, Bryant S, Kattine AA, et al. Cutaneous leiomyomas lack estrogen and progesterone receptor immunoreactivity. J Cutan Pathol. 1997;24:241–245.
26. Pithukpakorn M, Toro JR. Hereditary Leiomyomatosis and Renal Cell Cancer. In: Pagon RA, Bird TD, Dolan CR, Stephens K, eds. GeneReviews. Seattle, Washington: University of Washington; 1993–2006 Jul 31. [Updated 2010 Nov 02].
27. Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49–59.
28. Bunyan DJ, Eccles DM, Sillibourne J, et al. Dosage analysis of cancer predisposition genes by multiplex ligation-dependent probe amplification. Br J Cancer. 2004;91:1155–1159.
29. Naverson DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Dermatol. 1993;28:298–300.
30. Apatenko AK, Turusov VS. The painful tumours of the skin. Neoplasma. 1968;15:187–202.
31. Cook-Norris RH, Rodriguez AO, Kovach BT, et al. Segmental cutaneous piloleiomyomata. Skinmed. 2010;8:238–239.
32. Tiffee JC, Budnick SD. Multiple cutaneous leiomyomas. Report of a case. Oral Surg Oral Med Oral Pathol. 1993;76:716–717.
33. Englander L, Emer JJ, McClain D, et al. A rare case of multiple segmental eccrine spiradenomas. J Clin Aesthet Dermatol. 2011;4:38–44.
34. Montgomery H, Winkelmann RK. Smooth-muscle tumors of the skin. AMA Arch Derm. 1959;79:32–40.
35. George S, Pulimood S, Jacob M, Chandi SM. Pain in multiple leiomyomas alleviated by nifedipine. Pain. 1997;73:101–102.
36. Thompson JA Jr. Therapy for painful cutaneous leiomyomas. J Am Acad Dermatol. 1985;13:865–867.
37. Batchelor RJ, Lyon CC, Highet AS. Successful treatment of pain in two patients with cutaneous leiomyomata with the oral alpha-1 adrenoceptor antagonist, doxazosin. Br J Dermatol. 2004;150:775–776.
38. Thyresson HN, Su WP. Familial cutaneous leiomyomatosis. J Am Acad Dermatol. 1981;4:430–434.
39. Haugen RN, Tharp MD. The use of gabapentin for recurrent painful attacks with multiple piloleiomyomas. J Drugs Dermatol. 2008;7:401–402.
40. Sifaki MK, Krueger-Krasagakis S, Koutsopoulos A, et al. Botulinum toxin type A-treatment of a patient with multiple cutaneous piloleiomyomas. Dermatology. 2009;218:44–47.
41. Onder M, Adi?en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325–328.
42. Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319–322.
43. Aoki KR. Review of a proposed mechanism for the antinociceptive action of botulinum toxin type A. Neurotoxicology. 2005;26:785–793.
44. Aoki KR. Pharmacology and immunology of botulinum toxin type A. Clin Dermatol. 2003;21:476–480.
45. Hindley JT, Law PA, Hickey M, et al. Clinical outcomes following percutaneous magnetic resonance image guided laser ablation of symptomatic uterine fibroids. Hum Reprod. 2002;17:2737–2741.
46. Rongioletti F, Fausti V, Ferrando B, et al. A novel missense mutation in fumarate hydratase in an Italian patient with a diffuse variant of cutaneous leiomyomatosis (Reed’s syndrome). Dermatology. 2010;221:378–380.