
by Jo Ann LeQuang
Ms. LeQuang is a medical writer with LeQ Medical in Angleton, Texas.
This article is based on selected proceedings of the 2017 MauiDerm meeting, which was held March 20–24, 2017, at the Grand Wailea in Maui, Hawaii. Presentations included those by Brian Berman, MD; Roger I. Ceilley, MD; Linda Stein Gold, MD; Hayes B. Gladstone, MD; Arthur Kavanaugh, MD; Jim Krueger, MD, PhD; Craig L. Leonardi, MD; George Martin, MD; David M. Ozog, MD; Eggert Stockfleth, MD; Bruce E. Strober, MD, PhD; and Jashin J. Wu, MD.
J Clin Aesthet Dermatol. 2017;10(9 Suppl):S8–S41.
A message from the Guest Editor and MauiDerm Program Director:
George Martin, MD
Dermatology Associates, Kihei, Maui, Hawaii
Dear Colleagues:
This year’s MauiDerm 2017 supplement features several topics presented by our faculty that are relevant to your daily clinical practice. Given the rapid changes in the therapeutic landscape of dermatology, it is vital to stay abreast of new and novel therapies that can be both life changing and lifesaving to our patients. The following pages contain engaging scientific and clinical discussions of the latest advances in both psoriasis and cutaneous oncology, as well as case studies in challenging pediatric diseases.
I hope that you find the content both engaging and educational. We thank the educational sponsors of the MauiDerm meeting and this supplement for their commitment to dermatology and dermatology education.
With aloha,
George Martin, MD
MauiDerm 2017 Program Director; Guest Editor, the Journal of Clinical and Aesthetic Dermatology
Psoriasis
Psoriasis Update 2017
The UNVEIL study, a randomized, placebo-controlled, double-blind, multicenter, Phase 4 study ( NCT02425826) of the efficacy and safety of apremilast for the treatment of moderate plaque psoriasis was recently published.[1] Apremilast is a novel immunomodulatory medication approved for treatment of both psoriasis and psoriatic arthritis. It inhibits the activity of phosphodiesterase 4 (PDE4), which is active in immune system cells as well as in keratinocytes. Reducing PDE4 may decrease production of proinflammatory cytokines. To enroll in the study, adults (individuals older than 18 years) had to have chronic plaque psoriasis for at least six months prior to screening. Chronic plaque psoriasis was defined as a body surface area (BSA) of 5 to 10 percent and a score on the Static Physician’s Global Assessment (sPGA) of 3 on a scale of 0 to 5. The product of these two scores accordingly could range from 15 to 30 at screening and baseline. Patients were included when they had not previously been exposed to conventional systemic or biologic therapy for psoriatic arthritis, psoriasis, or any indication which might impact psoriasis assessment. The main exclusionary criterion was any inflammatory and/or dermatologic condition other than plaque psoriasis (including other forms of psoriasis). In the UNVEIL study, patients were randomized in a 2:1 scheme to receive apremilast or placebo for the first 16 weeks; at Week 16, placebo patients were switched to apremilast. All patients then continued on apremilast in open-label treatment until the end of the study (Week 52).
The primary efficacy endpoint was the mean percentage change from baseline at Week 16 in sPGA x BSA. The sPGA score was calculated for all plaques in all involved areas, erythema severity, scaling, and plaque elevation; these scores were then averaged and rounded to the nearest whole number. The secondary efficacy endpoints were the mean percentage change from baseline in the Psoriasis Area and Severity Index (PASI) score, the percentage of patients who could achieve at least a 75-percent reduction from baseline of the sPGA x BSA, as well as the percentage of patients who scored 0 (clear) or 1 (almost clear) in sPGA response with at least a two-point reduction from baseline.
A total of 221 patients were enrolled in the study. At 16 weeks, the mean percentage change of the sPGA x BSA was 48.1 percent for the apremilast group versus 10.2 percent of the placebo patients. sPGA x BSA that showed at least a 75-percent reduction over baseline was achieved by 35.1 percent of apremilast patients compared to 12.3 percent of placebo patients. About one-third of apremilast patients (30.4%) had a PGA score of 0 or 1 after 16 weeks, compared to 9.6 percent of placebo patients.
Adverse events occurred in 62.6 percent of apremilast and 47.9 percent of placebo patients, most of which were not serious or severe; three serious and three severe adverse events occurred in the apremilast group compared to one severe adverse event in the placebo group. The rate of study discontinuation owing to adverse events was 4.1 percent for placebo patients versus 3.4 percent for the apremilast group. The most frequently reported adverse effects were diarrhea (29.3% vs. 16.4% for apremilast vs. placebo, respectively), headache (20.4% vs. 11.0%), nausea (17.7% vs. 9.6%), upper respiratory tract infections (6.8% vs. 4.1%), decreased appetite (4.1% vs. 5.5%), and vomiting (6.1% vs. 2.7%).
Certolizumab pegol (CZP), infliximab, adalimumab, golimumab, and etanercept are five anti-tumor-necrosis-factor-alpha (anti-TNFalpha) drugs approved by the United States Food and Drug Administration (FDA) for treating psoriatic arthritis. At the late-breaking sessions of the 2017 American Academy of Dermatology Scientific Sessions, Gottlieb et al presented results of two randomized, double-blind, placebo-controlled, multicenter, 16-week, Phase 3 studies evaluating CZP in chronic plaque psoriasis patients: CIMPASI-1 (An Efficacy and Safety Study of Two Dose Levels of Certolizumab Pegol [CZP] in Subjects With Plaque Psoriasis [PSO] NCT02326298) and CIMPASI-2 (A Study to Evaluate the Efficacy and Safety of Two Dose Levels of Certolizumab Pegol [CZP] in Subjects With Plaque Psoriasis [PSO] NCT02326272).[2] Upon enrollment, patients were randomized in the 16-week, double-blind phase to placebo, CZP 200mg every two weeks (with a loading dose of CZP 400mg at Weeks 0, 2, and 4), or CZP 400mg every two weeks in a 1:2:2 ratio. The primary endpoints were the percentage of patients who, after 16 weeks of treatment, achieved a 75-percent reduction in their PASI score (PASI 75) and a PGA score of 0 (clear) or 1 (almost clear) on a 0-to-5 scale with at least a two-category improvement over baseline.
At 16 weeks, patients moved into the maintenance phase (Weeks 16–48) and placebo patients were migrated to the LD-CZP group. Nonresponders, defined as those with less than a PASI 50 score, had the opportunity to withdraw at Weeks 32, 40, and 48 and progress to the next group or “escape” to 400mg CZP every two weeks. At 48 weeks, all patients remaining in the study were administered CZP 200mg every two weeks in an open-label treatment phase, which ran to 144 weeks. A safety follow-up phase continued to Week 152.
There were significantly more responders in the CZP groups (both 200 and 400mg) versus placebo in terms of PASI 75 and PASI 90 rates, as well as PGA responder rates.
The CIMPACT study (Efficacy and Safety Study of Certolizumab Pegol [CZP] Versus Active Comparator and Placebo in Subjects With Plaque Psoriasis [PSO] NCT02346240) evaluated placebo, 200mg CZP, 400mgCZP (every two weeks for both doses), and etanercept 50mg twice weekly (for 12 weeks) as an active comparator. Patients were randomized in a 1:3:3:3 scheme (1 for placebo) to enter an initial, 16-week, double-blind treatment phase. The primary endpoint was PASI 75 at 12 weeks. After 16 weeks, patients entered the maintenance phase and from Week 48 to 144, followed by an open-label treatment phase. The CZP groups had significantly more PASI 75 and PASI 90 scores at Weeks 12 and 16 than placebo patients (Table 1).[3]

New and Emerging Psoriasis Treatments
From new studies on old drugs to innovative new agents, the systemic treatment of psoriasis remains in an exciting period of breakthroughs.
Methotrexate. A new, randomized, double-blind, placebo-controlled, Phase 3 clinical trial from Europe (METOP [Trial in Patients With Psoriasis Treated With Methotrexate Using an Optimized Treatment Schedule NCT02902861]) enrolled 149 methotrexate-naïve plaque psoriasis patients with moderate-to-severe disease.[4] Patients were randomized 3:1 to receive subcutaneous methotrexate (starting at 17.5mg/week with dose escalation in first eight weeks allowed up to a maximum of 22.5mg/week) or placebo over 16 weeks, then entered a maintenance phase of 36 more weeks, in which all patients were administered subcutaneous methotrexate. All patients received 5mg/week of folic acid throughout the study. By the 16th week, 41 percent of methotrexate and 10 percent of placebo patients had achieved a score of PASI 75 (relative risk 3.9, 95% confidence interval [CI]: 31–11.81, p=0.0026). In the methotrexate arm, 31 percent had their dose escalated to the maximum of 22.5mg/week. An sPGA score of 0 (clear) or 1 (almost clear) was achieved by 27 percent in the methotrexate and seven percent in the control groups. In this study, there were no patient deaths, no major adverse cardiac events (MACE), and no malignancies, but adverse events occurred in 82 percent of the methotrexate and 93 percent of placebo patients during the double-blind phase and then in 78 percent of methotrexate and 77 percent of placebo patients during the open-label phase. These findings contribute to the data about subcutaneous methotrexate and its potential efficacy and safety.
Second-generation biologics: Tumor necrosis factor-a antagonists. Biologics are the mainstay of systemic psoriasis treatment. Tumor necrosis factor (TNF)a antagonists include adalimumab, etanercept, infliximab, golimumab, and certolizumab. Biosimilars for infliximab, adalimumab, and etanercept have been approved or have been unanimously recommended to the FDA for approval.
Since psoriasis is inherently associated with increased cardiovascular risk, a retrospective study was conducted to evaluate the time to a MACE in patients receiving a TNF-alpha inhibitor versus those receiving methotrexate. Data from the first quarter of 2000 to the third quarter of 2011 were evaluated, and adult psoriasis patients with two or more prescriptions for either a TNF-alpha antagonist or methotrexate were included (n=17,729).[5] Overall, those who used TNF-alpha antagonists had a lower incidence of cardiovascular adverse events compared to methotrexate users (hazard ratio [HR]: 0.55, p<0.01); at 12 months; those using TNFalpha inhibitors had fewer cardiovascular events than those taking methotrexate (Figure 2). The time to first MACE among psoriasis patients showed that cumulative exposure to TNF-alpha blockade lessens cardiovascular risk over time compared to methotrexate.[5] The hazard reductions dropped by 21.3, 38.0, and 51.2 percent for one, two, and three years, respectively, of TNF-alpha blockade. Further, cumulative exposure to TNF-alpha blockade could be associated with an overall reduced risk for MACE.[5]
Interleukin-23 p19 subunit inhibitors. Tildrakizumab. Interleukin (IL)-23 p19 subunit inhibitors include tildrakizumab, guselkumab, and risankizumab. Reich et al reported results from the reSURFACE1 trial (A Study to Evaluate the Efficacy and Safety of Subcutaneous MK-3222, Followed by an Optional Long-Term Safety Extension Study, in Participants With Moderate-to-Severe Chronic Plaque Psoriasis [MK-3222-010] NCT01722331) and reSURFACE2 trial (A Study to Evaluate the Efficacy and Safety/Tolerability of Subcutaneous Tildrakizumab [SCH 900222/MK-3222] in Participants With Moderate-to-Severe Chronic Plaque Psoriasis Followed by a Long-term Extension Study [MK-3222-011] NCT01729754), demonstrating that tildrakizumab achieved rates of PASI 90 in almost 60 percent and PASI 100 in 24 percent of patients with moderate-to-severe plaque psoriasis who were treated for 28 weeks.[6] The reSURFACE2 study had four arms: tildrakizumab 200mg, tildrakizumab 100mg, etanercept 50mg, and placebo. At 12 weeks, placebo patients transitioned to one of the two tildrakizumab groups and remained there until Week 28. Compared head-to-head, tildrakizumab offered significant improvements in the percentage of patients achieving PASI 75 versus both placebo (p<0.001) and etanercept (p<0.05), and while PGA scores were significantly better for tildrakizumab versus placebo, the difference achieved statistical significance versus etanercept only for tildrakizumab 200mg (p<0.05), but not the 100mg dose.[6]
The proportion of patients having at least one adverse event in the reSURFACE2 study was highest in the placebo group (55.1%), followed by etanercept (54.0%) and tildrakizumab 100 and 200mg (44.3% and 49.4%, respectively). Adverse events culminating in withdrawal from the study happened in less than two percent of patients in all groups (highest in etanercept group at 1.9%, lowest in both tildrakizumab groups at 1.0%, 1.3% dropout rate in placebo patients). The most commonly reported adverse events were injection-site erythema and nasopharyngitis.
Guselkumab. The results of the VOYAGE 1 trial (A Study of Guselkumab in the Treatment of Participants With Moderate to Severe Plaque-Type Psoriasis NCT02207231), a Phase 2, randomized, double-blind, placebo- and active-comparator-controlled study, were presented in 2016 and recently published.[7] Psoriasis patients were randomized to be treated with guselkumab 100mg (at Weeks 0 and 4, then every 8 weeks thereafter), adalimumab (80mg at Week 0, 40mg at Week 1, then 40mg every 2 weeks), or placebo for 16 weeks, then guselkumab for the open-label phase. Compared to placebo, a significantly greater proportion of guselkumab patients scored 0 (clear) or 1 (minimal) on the five-point Investigator Global Assessment (IGA) score at 16 weeks (85.1% vs. 6.9%, respectively, p<0.001).[7] A greater proportion of guselkumab patients scored PASI 90 compared to placebo patients (73.3% vs. 2.9%). Compared to adalimumab, significantly greater proportions of guselkumab patients scored 0 or 1 on the IGA at Weeks 16, 24, and 48 (85.1% vs. 65.9%, 84.2% vs. 61.7%, and 80.5% vs. 55.4%, respectively) and significantly more guselkumab patients achieved PASI 90 at Weeks 16, 24, and 48 (73.3% vs. 49.7%, 80.2% vs. 53.0%, and 76.3% vs. 47.9%, respectively). The VOYAGE2 trial (A Study of Guselkumab in the Treatment of Participants With Moderate to Severe Plaque-Type Psoriasis With Randomized Withdrawal and Retreatment NCT02207244)8 reported better persistence of response in the guselkumab maintenance group versus the withdrawal arm of the study (p<0.001) from Weeks 28 to 48. About two-thirds of the adalimumab nonresponders (66.1%) who were switched to guselkumab at Week 28 achieved PASI 90 at Week 48. The rate of adverse events was similar among groups for both VOYAGE1 and VOYAGE2.
Risankizumab. Risankizumab is a novel anti-IL-23 monoclonal antibody (BI 655066) that was recently studied for safety, efficacy, and pharmacokinetic properties in a first-in-human, proof-of-concept study, designed as a single-rising-dose, randomized, double-blind, placebo-controlled trial.[9] Eighteen patients with moderate-to-severe plaque psoriasis received 0.01, 0.05, 0.25, 1, 3, or 5mg/kg of risankizumab intravenously (IV), 0.25 or 1mg/kg risankizumab subcutaneously (n=13), or matched placebo (n=8). Adverse events were similar in the active and control groups. Risankizumab was associated with clinical improvement commencing around Week 2 and maintained by 33 percent of those treated up to 66 weeks after treatment. PASI 75, PASI 90, and PASI 100 scores were achieved at 12 weeks by 87, 58, and 16 percent of risankizumab patients, respectively; no placebo patients scored in this range.[7] As such, risankizumab may be considered an “immunologic disrupter.”
Third-generation biologics. IL-17 inhibitors. IL-17 inhibitors include secukinumab, ixekizumab, and brodalumab. There are clear differences in these three agents in terms of structure and signaling,10 although all three may be considered next-generation biologics (Figure 1).

Secukinumab. The SCULPTURE study (Efficacy and Safety of Subcutaneous Secukinumab [AIN457] for Moderate to Severe Chronic Plaque-type Psoriasis Assessing Different Doses and Dose Regimens [SCULPTURE] NCT01406938), presented at the European Academy of Dermatology and Venereology Congress in Vienna in 2016 by Bissonnette et al, demonstrated durable effectiveness of secukinumab in a four-year observed analysis.[11] At the end of Year 4 (n=131), 88.5, 66.4, and 43.5 percent of secukinumab patients had PASI 75, PASI 90, and PASI 100 scores, which were similar to first-year results (n=162) of 88.9, 68.5, and 43.8 percent, respectively. The rate of treatment-emergent adverse events remained similar over the years (78.0% at Year 1, 75.0% at Year 2, 69.4% at Year 3, 62.0% at Year 4) with the most frequently reported adverse events being nasopharyngitis, upper respiratory tract infection, headache, and pharyngitis. Neither MACE nor opportunistic infections (other than candida infections) occurred in four years.[11]
Ixekizumab. Ixekizumab has been the focus of numerous recently published studies.[12] The long-term results from a Phase 2 study (A Study in Participants With Moderate to Severe Psoriasis NCT01107457) of five-year safety and efficacy were recently presented at the American Association of Dermatology (AAD) scientific sessions. Subcutaneous ixekizumab was administered six times at Weeks 0, 2, 4, 8, 12, and 16; patients with plaque psoriasis for six or more months involving at least 10 percent of BSA then entered a treatment- free period up to Week 32; those whose response was better than PASI 75 at Week 32 were eligible to enter the open-label extension phase. In this phase, patients received 120mg of subcutaneous ixekizumab every four weeks up to Week 88, and then 80mg of subcutaneous ixekizumab every four weeks thereafter to Week 240 (data on file with Eli Lilly and Company).
Adverse events caused 10.8 percent of reported adverse events included alanine aminotransferase increase, hepatic enzyme increase, hidradenitis, invasive ductal breast carcinoma, nonsmall-cell lung metastatic cancer, osteomyelitis, psoriatic arthropathy, pyelonephritis, rectal adenocarcinoma, rectal adenoma, or a urinary tract obstruction (all <1%) or due to pregnancy (1.7%). No deaths were reported. Treatment-emergent adverse events decreased from 60.0 percent in the first year to 50.0 percent in the fourth year, but serious adverse events increased from 5.8 percent in the first year to 10.5 percent in the fourth year. Serious infections occurred in 5.0 percent of patients.
Brodalumab. In two Phase 3 studies (the AMAGINE2 (Study of Efficacy and Safety of Brodalumab Compared With Placebo and Ustekinumab in Moderate to Severe Plaque Psoriasis Subjects NCT01708603) and AMAGINE3 (Study of Efficacy and Safety of Brodalumab Compared With Placebo and Ustekinumab in Moderate to Severe Plaque Psoriasis Subjects NCT01708629), patients with moderate-to-severe psoriasis were randomized into one of four groups: brodalumab 210mg every two weeks, brodalumab 140mg every two weeks, ustekinumab 45mg or 90mg depending on patient weight, or placebo.[13] After 12 weeks, brodalumab patients were randomized again to a maintenance dose of brodalumab of either 210mg every two weeks, 140mg every two weeks, 140mg every four weeks, and 140mg every eight weeks. Ustekinumab patients continued to receive their regimen unchanged, while placebo patients were migrated to the 210mg brodalumab group. At 12 weeks, PASI 75 response was higher in both 210mg and 140mg brodalumab groups (86% and 67%, respectively) than placebo (8%), p<0.001. The number of brodalumab patients with sPGA scores of 0 and 1 was significantly higher than placebo patients (p<0.001). At 12 weeks, PASI 100 scores were achieved by significantly more brodalumab 210mg patients than ustekinumab patients in both studies (44% vs. 22%, respectively, and 37% vs. 19%, respectively, for AMAGINE2 and AMAGINE3, p<0.001 for both). For adverse events, neutropenia was more common with active treatments than placebo, and mild-to-moderate candidate infections occurred more often in brodalumab patients than ustekinumab or placebo patients.[13]
Safety concerns have arisen about brodalumab and its possible associations with depression and suicidality. In Phase 3 trials, the drug sponsor, Amgen (Thousand Oaks, California), introduced a psychological survey instrument to better assess patients who might be at risk for these outcomes, but the intervention was not successful in identifying patients at risk. Despite robust efficacy results, studies were halted and Amgen stopped developmental activities in the second quarter of 2015. Amgen’s partners continued development and submitted brodalumab to the FDA where it was reviewed in July of 2016. At the Dermatologic and Ophthalmic Drugs Advisory Committee (DODAC) on July 19, 2016, 18 panelists recommended approval and 14 recommended a risk evaluation and mitigation strategy program (REMS) for brodalumab. Thus, brodalumab was approved in February 2017 with a black-box warning, and a REMS program was established to restrict access. Patients who are prescribed brodalumab must be counseled and made aware of the mental health risks, and any new or worsening symptoms of depression or suicidality must be reported and patients will be referred, as appropriate, to competent mental health professionals. Patients must sign a patient-prescriber agreement in order to take brodalumab, which only certified pharmacies may dispense. Brodalumab is contraindicated in patients with Crohn’s disease.[14]
It has long been reported in the literature that psoriasis patients are at elevated risk for suicidality apart from any particular course of treatment. In a 1993 study, a survey of 217 psoriasis patients revealed that 9.7 percent said they had a wish to be dead and 5.5 percent had active suicidal ideation at the time of the survey.[15] These traits could be associated with a higher score for depression (p<0.0001) and higher self-ratings of severity of psoriasis (p<0.05). This important association between psoriasis and suicidality plus the potential risk factors specifically associated with brodalumab remain to be further elucidated.
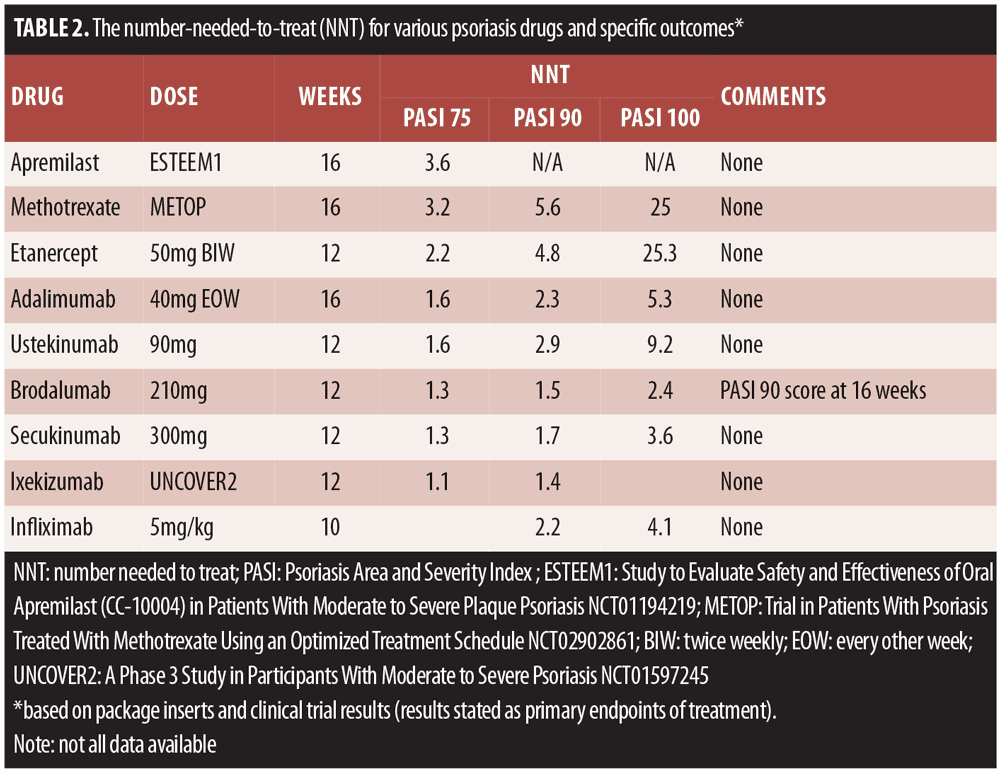
Treatment targets. The AAD published treatment targets for plaque psoriasis in 2017 that recommended BSA as the preferred assessment metric and that treatments ought to attain an improvement of at least 75 percent every three months or the equivalent of achieving three percent BSA improvement every three months.[16] The target response would be less than one percent of BSA achieved at six months and then maintained every six months thereafter.[16] In order to meet this goal, dermatologists must select highly effective drugs for their psoriasis patients. An important metric for appropriate drug selection is the number needed to treat (NNT), which defines the average number of patients who must be treated to achieve one additional good outcome. In clinical trials, the NNT is the number of patients who are treated to get one patient to benefit when compared to a control patient.[17] Fortunately, based on prescribing information provided in product labeling and clinical trials, it is possible to know the NNT scores for a variety of psoriasis drugs for various outcomes (Table 2).
The old treatment paradigm for psoriasis modeled a stepwise progression starting with over-the-counter (OTC) products, such as emollients, and advancing to prescription topicals (including topical steroids, vitamin D analogs, retinoids), phototherapy options, and finally systemic therapy. In this traditional treatment scheme, patients had to move step by step through the system and could not advance to the next step until they had failed the current step. In other words, patients could not get phototherapy until they had failed prescription topicals. This has given way to our emerging psoriasis treatment model, which commences with topical therapy; if a patient fails topical therapy (OTC or prescription products), the patient may be advanced to phototherapy, traditional systemic agents (such as apremilast, acitretin, methotrexate), or biologics, based on the individual patient’s preferences and condition.
Psoriatic Arthritis Update
The societal costs of psoriatic arthritis (PsA) have been and remain enormous. In a nationwide cohort study from Denmark using data from January 1998 through December 2014, 10,525 PsA patients were matched with 20,777 comparators from the general population.[18] At baseline, compared to the general population, PsA patients had more comorbid conditions: cardiovascular disease (odds ratio [OR] 1.70, 95% CI: 1.54–1.6), respiratory disease (OR 1.73, 95% CI: 1.54–1.96), and infectious diseases (OR 2.03, 95% CI: 1.69–2.42). PsA patients had higher total healthcare costs and significantly lower income than people in the general population. Perhaps most concerning is the fact that five years prior to PsA diagnosis, PsA patients were at greater risk of being on disability (risk ratio [RR] 1.36, 95% CI: 1.24–1.4) than individuals in the general population, but that risk increased to 1.60 (95% CI: 1.49–1.72) at the time of diagnosis, and then increased to RR 2.69 (95% CI: 2.40–3.02) 10 years after diagnosis. In fact, a decade after the PsA diagnosis, 22 percent of PsA patients were on disability insurance.[18]
About 2 to 3 percent of the population has cutaneous psoriasis, and, of that population, about 30 percent will develop PsA.[19] Thus, it becomes important to the healthcare system to better identify those psoriasis patients who are most at risk of developing PsA, to allow for earlier and potentially more effective intervention. The question remains unanswered as to why some patients with psoriatic skin disease do not develop PsA. It has been speculated that PsA may exist in all patients with cutaneous psoriasis but it remains subclinical in most. Better understanding of this so-called “occult PsA” and the transition from psoriatic skin disease to PsA will allow for development of better therapeutic targets. In a study of 85 subjects (55 psoriasis patients and 30 healthy control subjects), high-field magnetic resonance imaging (MRI) scans were used to evaluate patients with psoriasis for synovitis, osteitis, tenosynovitis, and periarticular inflammation based on the PsA MRI scoring (PsAMRIS) criteria.[10] At least 47 percent of these patients had at least one inflammatory lesion visible on MRI. The frequencies of the observed lesions were 38 percent inflammatory, 11 percent osteitis, four percent tenosynovitis, and four percent periarticular inflammation. In psoriasis patients with subclinical synovitis and arthralgia-associated symptoms, the risk for developing PsA was as high as 55.5 percent, but was 15.3 percent for those patients with normal MRI scans and no reported arthralgia symptoms.[20]
C-C-X motif ligand (CXCL) 10 is a chemokine which is secreted by a variety of cells, including lymphocytes, monocytes, keratinocytes, fibroblasts, and endothelial cells in response to interferon-gamma (IFNgamma) and TNF.[21] It has been speculated that CXCL10 may be a potential biomarker for PsA. In a prospective study of patients with skin psoriasis but without PsA, baseline levels of serum CXCL10 messenger RNA were evaluated as a potential biomarker for PsA.22 Patients in whom PsA developed were designated in this study as “converters,” compared to “nonconverters” (who did not develop PsA). CXCL10 serum levels were significantly higher in converters (493pg/mL) than nonconverters (371pg/mL), p=0.005, but serum levels of C-reactive protein were similar between groups.[22] After PsA developed, patients exhibited lower levels of CXCL10. This both implicates CXCL10 as a predictive biomarker for PsA in psoriasis patients and suggests that it may play a role in the pathogenesis of PsA.
A new population-based study by Lewinson et al[23] has found depression increases the risk that a patient with cutaneous psoriasis will develop PsA. Since systemic inflammation increases the risk of PsA in psoriasis patients and since major depressive disorder (MDD) is associated with increased levels of systemic inflammation, investigators hypothesized that patients with psoriasis who suffer from MDD were more at risk of developing PsA than those without MDD. A retrospective analysis using the Health Improvement Network identified psoriasis patients (n=73,447) who were followed for 25 years until either PsA was diagnosed or the censor date. Using Cox proportional hazards models, those psoriasis patients who developed MDD had a significantly increased risk of subsequently developing PsA compared to those psoriasis patients who did not have MDD (HR: 1.37, 95% CI: 1.05–1.80, p=0.021).[23]
There are numerous treatment options for PsA, including adjunctive treatments (e.g., nonsteroidal anti-inflammatory drugs [NSAIDs]), physical therapy, topical agents (e.g., topical corticosteroids), disease-modifying antirheumatic drugs (DMARDs) (e.g., methotrexate), biologics, and new experimental agents (e.g., Janus-kinase [JAK] inhibitors, alpha-IL-23 monoclonal antibodies, and other anti-IL-17 agents).
Early and effective treatment of PsA is crucial to reduce joint damage, improve outcomes, prevent disability, and enhance patient quality of life. As many as 20 to 50 percent of patients with PsA are unemployed and 16 to 39 percent are legally disabled.[24] Patients with PsA who are employed may suffer from presenteeism (i.e., ability to go to work but at a diminished capacity), absenteeism (i.e., taking time off from work for medical reasons), and productivity loss (i.e., combination of presenteeism plus abstenteeism).[25] A multicenter, observational cohort study enrolled patients with PsA who were either starting or switching to an anti-TNF therapy or conventional synthetic DMARD (csDMARD) for PsA.[25] At baseline, 57 percent of the study population was employed and 24 percent of those of working age were not employed. At six months of therapy, there were significant improvements in the csDMARD group (n=164) in terms of presenteeism (10%, p=0.007) and work-related productivity (15%, p=0.001). The improvements were more pronounced in the anti-TNF therapy group (n=65), with proportions of patients achieving significantly less presenteeism (30%, p<0.001) and greater work productivity (40%, p<0.001). Clinical improvement was greater in the anti-TNF group compared to the csDMARD group.[25]
Obesity is a frequently observed comorbidity with PsA and may exacerbate its course.[27] Obesity is associated with inflammation, and studies of other rheumatic patients suggest that anti-TNF therapy adherence is low in patients with obesity.[26,27] An observational cohort study evaluated patients with PsA over a median 1.5 years (n=1,943).[28] Of this population, 32 percent were obese (n=408), defined as body mass index (BMI) of 30kg/m2 or more. Adherence was defined as the number of years that the patient maintained treatment with the first TNF inhibitor, although temporary interruptions in treatment (<3 months) were allowed. TNF inhibition therapy was shorter in patients with obesity compared to patients without obesity (2.5 years vs. 5.9 years). Good or moderate response on the European League Against Rheumatism (EULAR) scale was achieved by 55 percent of obese and 65 percent of nonobese patients at six months (p=0.02), and obesity was associated with greater degree of disease activity, increased risk of discontinuation of TNF inhibition therapy, and decreased treatment response. Obesity had a significant effect on patients in this study irrespective of sex, particular type of TNF inhibitor, and patient nationality.[28]
Biologics able to inhibit the IL-17A pathway have been explored for their potential role in treating PsA as well as other disorders. IL-17A has been implicated in the pathogenesis of multiple immunoinflammatory diseases, including psoriasis, PsA, and rheumatoid arthritis (RA).[29] IL-17 is a cytokine family of six subunits (A, B, C, D, E, F). IL-17A may be described as a principal effector of T-helper type (Th) 17 cells; high levels may be found at diseased cutaneous sites and in diseased joints.[29] In results presented at the 2016 meeting of the American College of Rheumatology, secukinumab at multiple dosages offered sustained improvements in signs and symptoms in various clinical domains at three years in 606 PsA patients.[30]
Other IL-17A inhibitors currently being evaluated for PsA treatment include ixekizumab, bimerkizumab (this monoclonal antibody antagonizes both IL-17A and IL-17F), a dual-variable domain (DVD) drug that inhibits TNF and IL-17 (ABT-122), and guselkumab. A novel selective T-cell costimulator (abatacept) used to treat rheumatoid arthritis and juvenile idiopathic arthritis was recently evaluated in the ASTRAEA trial (Efficacy and Safety of Subcutaneous Abatacept in Adults With Active Psoriatic Arthritis [ASTRAEA] NCT01860976) and was shown to be effective against PsA and well tolerated, even in patients previously exposed to TNF inhibitors.[31]
New agents that help to inhibit JAK pathways are being explored for their potential role in treating psoriasis and rheumatic diseases.[32] The two best known JAK inhibitors are tofacitinib and ruxolitinib. In a randomized placebo- and active-controlled, double-blind, Phase 3 study, PsA patients were randomized to one of five groups: oral tofacitinib 5mg twice daily (n=107), tofacitinib 10mg daily (n=104), subcutaneous adalimumab 40mg once every two weeks (n=106), placebo patients who were migrated to tofacitinib 5mg twice daily at the end of three months (n=52), and placebo patients who were migrated at three months to 10mg of tofacitinib twice daily (n=53).[33] Tofacitinib was significantly superior to placebo on the American College of Rheumatology and 20-percent improvement (ACR20) response rates and improved scores on the Health Assessment Questionnaire Disability Index at three months. Safety was similar in all groups.
The OPAL BEYOND trial (Tofacitinib In Psoriatic Arthritis Subjects with Inadequate Response to TNF Inhibitors NCT01882439) examined tofacitinib’s effectiveness in 395 patients with PsA who did not respond to TNF inhibition therapy.[34] Effectiveness with tofacitinib was superior to placebo in ACR20 response and improved scores on the Health Assessment Questionnaire Disability Index with superiority becoming evident as early as the first assessment (Week 2). No new safety risks emerged in this study.
Topical Therapies for Psoriasis
The vehicle for topical products plays a large role in effectiveness, safety, and patient satisfaction. For example, topical calcipotriene produced marked improvement in more patients (70%) as an ointment compared to solution (31%), foam (41%), or cream (50%). However, skin irritation is markedly lower in the solution and foam formulations; calcipotriene ointment and cream have rates of skin irritation around 10 to 15 percent. This gives rise to the question as to whether “designer vehicles” can be created to help optimize drug penetration into the skin, skin permeation rates, and how well the active ingredients are absorbed into the receptor fluid.
To this end, a novel betamethasone dipropionate (BD) emollient spray was formulated, aimed at gaining better epidermal penetration in skin affected by psoriasis while minimizing systemic absorption. This new formulation was compared with super-potent augmented BD 0.05% lotion in adults with moderate plaque psoriasis.[35] Patients in this study (n=351) were randomized to receive augmented BD or the new emollient spray or vehicle alone to be applied twice a day to affected areas for two weeks. The primary endpoint was an IGA score of 0 or 1 and at least two-grade improvement over baseline. At baseline, the mean BSA affected by psoriasis in patients was about 13 percent. The primary endpoint was achieved at two weeks in 19.0 percent of the emollient spray patients, 18.9 percent of the superpotent augmented BD lotion patients, and 2.3 percent in the vehicle patients (spray was significantly better than placebo, p<0.001). Response was rapid, with 10 percent of patients responding to the emollient spray at eight days versus 6.7 percent of augmented BD patients and 1.2 percent of vehicle patients. All products were well tolerated but significantly more patients reported burning or stinging with augmented BD compared to the emollient spray (13.6% vs. 4.1%, p=0.006).[35]
Combination therapy may be an important consideration in topical therapy of psoriasis. The rationale behind combination therapy is to enhance efficacy by using multiple products with complementary mechanisms of action while, when possible, reducing side effects by using lower doses of each medication.[36] For example, topical combination therapies have been proposed with the aim of reducing steroid use while still providing safe, effective treatment. In a three-arm study of calcipotriene ointment 0.005% applied mornings and halobetasol propionate 0.05% ointment applied evenings, this combination therapy was compared to the twice-daily use of either product alone (n=127).[37] At two weeks, the calcipotriene/halobetasol combination group had significantly improved results in the treatment of psoriasis and fewer cutaneous adverse effects than either of the two components applied in monotherapy. Lesional or perilesional irritation, a side effect sometimes reported with calcipotriene ointment monotherapy, did not occur in the combination group.[37] This study terminated at two weeks. To assess the durability of therapies, a double-blind, placebo-controlled study of the 40 patients in the prior study who achieved 50-percent or greater improvement were then randomized to receive either halobetasol ointment twice daily on weekends and calcipotriene ointment twice daily on week days (n=20) or halobetasol ointment twice daily on weekends and placebo ointment twice daily on weekdays (n=20). At six months, 66 percent of the combination patients were able to maintain remission compared to 40 percent of the control patients.[38]
The drawback with combination therapy that relies on two different products is that it may be too complex, inconvenient, or burdensome for patients to manage self-application over the long term. A fixed combination product, which combines calcipotriol and BD into a single ointment, can be applied once daily. In a clinical trial of 1,603 patients with psoriasis randomized to one of four treatment arms (combination, betamethasone only, calcipotriol only, or vehicle), the mean percentage change in PASI scores at the end of four weeks of treatment was -71.3 for combination therapy compared to -57.2, -46.1, and -22.7 for betamethasone monotherapy, calcipotriol monotherapy, or vehicle, respectively.[39] The mechanism of action behind this combination calcipotriol/BD product is thought to be based on the fact that the combination suppresses both TNFalpha and the IL-23/IL-17 axis more effectively than BD monotherapy.[40]
The ability of a topical product to penetrate the stratum corneum may improve its bioavailability. The only substance that can penetrate the skin barrier is the dissolved active ingredient (not crystalline substances), and the rate of cutaneous penetration is proportional to the amount of dissolved active ingredient. Thus, if the solubility of active ingredients could be improved and delivered in a vehicle that patients preferred, this could increase not just the drug’s usability but also its clinical effectiveness. The efficacy of the fixed-combination calcipotriene/BD product in an aerosol foam formulation compared to an ointment was studied in a multicenter, prospective randomized trial (n=376); this study was investigator-blinded only (not double-blinded) as there were formulation differences that could not be masked. At four weeks, psoriasis patients had significantly greater improvement with the combination foam product (54.6%) compared to combination ointment (41.0%, p=0.025) and vehicles (foam 6.1%, ointment 7.8%).[41]
Tazarotene is an effective topical retinoid available in 0.1% and 0.05% cream and gel formulations. Side effects, including pruritus and erythema, are reported in up to 30 percent of patients. Combining tazarotene with topical steroids has long been observed to enhance efficacy while reducing potential adverse events.[42] A fixed-combination product of halobetasol propionate lotion 0.01% together with tazarotene 0.045% (HP/TAZ) was evaluated in the treatment of moderate-to-severe plaque psoriasis.43 In a randomized, double-blind, vehicle-controlled, Phase 2 study of 212 patients with moderate-to-severe psoriasis, subjects were randomized to one of four groups: HP/TAZ, HP monotherapy, TAZ monotherapy, or vehicle.[43] All products were applied once a day for eight weeks. Efficacy was at least two-grades improvement over baseline in IGA score and a score of “clear” or “almost clear.” At two weeks, HP/TAZ was statistically significantly superior to vehicle, and by the conclusion of the study, 52.5 percent of the combination patients (HP/TAZ) had achieved treatment success compared to HP only (33.3%), TAV only (18.6%), and vehicle (9.7%) groups. Adverse events most frequently reported were application-site reactions.[43] The proportion of patients reporting at least one adverse event was 33.9, 21.0, 46.6, and 22.6 percent for HP/TAZ, HP monotherapy, TAZ monotherapy, and vehicle, respectively. A related Phase 3 study was completed that randomized 215 patients with moderate-to-severe plaque psoriasis to a combination lotion (halobetasol propionate 0.01% combined with tazarotene 0.045%) versus vehicle with primary outcomes defined as two-grade improvement or more from baseline in IGA score and an IGA score of 0 (clear) or 1 (almost clear). Results indicated that the combination product was significantly more effective than the vehicle (results on file with LS Gold, a presenter at MauiDerm 2017 whose presentation is one of which portions of this article are based). Overall, the combination product was well tolerated.
The forefront of topical research involves new molecules able to disrupt the many and varied intracellular signal-transduction pathways used by cytokines. Cytokines, chemokines, antibodies, and antigens bind to receptors on the surface of the cell, causing the receptors to polymerize, which enables their auto-activation or allows them to recruit binding partners.[44] This, in turn, results in a cascade of signaling activity ,which lead to the altered expression of the genes involved in inflammation, degradation of the extracellular matrix, apoptosis, and other cellular processes. Many of these signaling pathways are currently known, and more are being discovered. Among the pathways known to play a role in psoriasis are the mitogen-activated protein kinase (MAPK) pathway, the spleen-tyrosine kinase (Syk) pathway, the phosphoinositide 3-kinase (P13K0 pathway), the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) pathways, the protein kinase C (PKC) pathway, the nuclear-factor-kappa light-chain-enhancer of activated B-cells (NF-kappaB) pathway, and, perhaps most discussed currently, the JAK pathway.[44,45]
A cytokine binding to a receptor can activate JAK, a tyrosine kinase. Activated JAK acts as a docking station for signal-transducer and activator of transcription (STAT) factor, which induces pro-inflammatory cytokine gene transcription. Once the STAT is docked, it is phosphorylated by the activated receptor-associated JAK. There are four JAK enzymes (JAK1, JAK2, JAK3, JAK4) that pair up in various combinations to integrate signaling from almost 40 different cytokines and growth factors.
When cytokines bind to the cell-surface receptors, the JAKs dimerize and are autophosphorylated. STAT proteins are bound and are phosphorylated by the activated JAKs. Phosphorylated STAT proteins then are translocated to the nucleus where they initiate gene transcription and secretion of the specific cytokine that activated the receptor. When the signaling processes are disrupted, some pro-inflammatory cytokines may be overproduced; increased cell proliferation may occur; and there may be an increase in multiple cellular processes, including growth/maturation of lymphoid cells and the differentiation/ homeostasis of T-cells and B-cells.[46–50]
Tofacitinib is a new small molecule that can be applied in an ointment formulation for the treatment of plaque psoriasis based on the inhibition of JAK1/JAK3. In a randomized, double-blind, parallel-group, vehicle-controlled Phase 2b study over 12 weeks, tofacitinib ointment in two strengths (2% and 1%) was applied once or twice daily in adults with mild-to-moderate plaque psoriasis.[51] A total of 430 patients were treated; tofacitnib ointment 2% applied once or twice a day was associated with greater effectiveness than the vehicle at Week 8 but not Week 12; about half of patients (44.2%) experienced adverse events but the higher rate of adverse events occurred in the vehicle rather than active-treatment group.[51]
A novel agent, known currently as INCB018424, is a powerful JAK1/JAK2 inhibitor that is able to block signal transduction of several pro-inflammatory cytokines. Topical INCB018424 phosphate 1% or 1.5% cream was applied once or twice daily for four weeks over 2 to 20 percent of the BSA in five sequential cohorts of five patients each. Target lesions were rated on a scale of 0 to 4 for erythema, scaling, and thickness; the Physician’s Global Assessment was also used to assess overall disease activity. Both formulations improved lesion scores.[52]
Another new small module, GSK2894512, has undergone a dose-finding Phase 2 study set up as a double-blind, vehicle-controlled, six-arm, parallel-group trial. More small molecules will likely be tested soon for topical delivery.
Update on Psoriasis Comorbidities
Psoriasis is associated with many comorbidities that may affect prescribing considerations. It has been known that psoriasis is comorbid with many cardiometabolic conditions (e.g., atherosclerosis, atrial fibrillation, diabetes, dyslipidemia, hypertension, metabolic syndrome, obesity, and peripheral vascular disease) as well as noncardiovascular conditions (e.g., anxiety, asthma, chronic kidney disease, chronic obstructive pulmonary disorder, depression, migraines, multiple sclerosis, and uveitis).[53] New and ongoing research continues to associate other conditions with psoriatic disease.
A family history of cardiovascular disease may predict MACE in patients with psoriasis. In a retrospective study from data drawn from the Danish National Patient Register (n=2,722,375),54 investigators found that patients who had both psoriasis and a family history of cardiovascular disease had an adjusted incidence rate ratio for MACE of 1.28 for mild disease (95% CI: 1.12–1.46) and 1.62 for severe disease (95% CI: 1.14–2.30). Patients with psoriasis of any degree of severity but without a family history of cardiovascular disease had no incremental risk for MACE.
Abnormalities in liver enzyme levels are commonly reported in patients with psoriasis.55 To investigate a potential association between psoriasis and autoimmune hepatitis, a retrospective Danish nationwide cohort study (n=5,404,503) observed 56,739 people with mild and 10,909 with severe psoriasis.56 The adjusted incidence rate ratio for autoimmune hepatitis was 2.64 (95% CI: 1.70–4.11) and 3.05 (95% CI: 0.98–9.47) for mild and severe psoriasis, respectively (p<0.001 for mild psoriasis vs. reference population, p=0.054 for severe psoriasis vs. reference population).[56]
Chronic pancreatitis may develop following episodes of acute pancreatitis. It is an irreversible inflammatory disease of the pancreas and confers a risk of morbidity and mortality.[57] A retrospective analysis of the Taiwan National Health Insurance Research Database (n=48,430 people with psoriasis and n=193,720 without psoriasis) found the incidence of chronic pancreatitis to be 0.61 per 1,000 person-years in patients with psoriasis compared to controls over a mean of 6.6 years.[58] Patients with mild psoriasis had significantly higher risk of chronic pancreatitis (crude hazard ratio 1.81, 95% CI: 1.53–2.15), and the risk appears to be similar with severe psoriasis (crude hazard ratio 1.68, 95% CI: 1.02–2.89). Data in this study were analyzed two ways: once including patients with psoriasis and PsA and once excluding the patients with PsA from the psoriasis group. Results did not differ significantly.
Avascular necrosis (AVN), sometimes called osteonecrosis, is an ischemic bone necrosis that can occur when blood supply to the bone is lost or diminished. In a retrospective study based on the Taiwanese National Health Insurance Research Database, 28,268 patients with psoriasis were identified and then matched to 113,072 randomly selected controls without psoriasis.[59] Patients with psoriasis had higher adjusted hazard ratio for AVN than control patients (HR: 1.96, 95% CI: 1.62–2.38), and men with psoriasis had significantly higher adjusted hazard ratio for AVN than women. The risk for AVN occurs in both mild and severe psoriasis disease (although severe psoriasis confers a greater risk than mild psoriasis) and in patients with and without PsA (although those with PsA had a greater risk for developing AVN). The risk for AVN was greater in patients under age 30 years than other age groups (adjusted HR 2.64, 95% CI: 1.24–5.61).
Endothelial dysfunction, considered an early phase of atherogenesis, is a systemic and pathological state of the endothelium that results from an imbalance between vasodilating and vasoconstricting substances produced by and/or acting on the inner lining of blood vessels. Accelerated atherosclerosis has been recognized as a comorbidity of moderate to severe psoriasis.[60, 61, 62] A new study explored whether the use of TNFalpha inhibitors in patients with moderate-to-severe psoriasis (n=29 consecutive patients) would improve endothelial function measured by brachial artery reactivity measuring flow-mediate dilatation (FMD%).[63] Patients had completed six months treatment with adalimumab. FMD% values increased (improved) from 6.19±2.44 percent at the commencement of adalimumab therapy to 7.46±2.43 percent after six months (p=0.008).
Arterial stiffness is considered an independent predictor of cardiovascular disease, and it was likewise found that after six months of treatment with adalimumab, patients with moderate-to-severe psoriasis showed significant improvement. Carotid arterial stiffness was assessed using pulse wave velocity (PWV) at baseline and then at six months. PWV decreased (improved) from 6.28±1.04m/sec at baseline to 5.69±1.31m/sec at six months (p=0.03).[63]
In a single-center, prospective, observer-blinded, controlled study, noncontrast coronary artery calcium computed tomography (CT) scans along with contrast-enhanced coronary CT angiography were performed at baseline and again at 13 months in 28 patients with severe psoriasis treated with adalimumab, etanercept, infliximab, or ustekinumab.[64] Patients could rotate to different drugs from this list over the 13 months of the study at the discretion of their physicians in order to control inflammation. Noncontrast coronary artery calcium scores remained stable in the active-treatment group but progressed in the control group (p=0.02). Luminal narrowing remained unchanged in the intervention group but worsened in the control group.[64] Thus, these biologics could be associated with a reduction in coronary artery disease progression in patients with severe psoriasis.
To compare the risk of MACE in patients treated with TNFalpha inhibitors versus those treated with methotrexate, a retrospective claims-based analysis was conducted using data from the Truven Health Analytics MarketScan® databases with data from the first quarter of 2000 through the third quarter of 2011).[65] In this large study (n=382,059 psoriasis patients), TNFalpha inhibition was associated with greater reduction of various cardiovascular risks in psoriasis patients than methotrexate (Figure 2, Table 3).


The Central Importance of the IL-23/Th17 Pathway in the Pathogenesis and Treatment of Psoriasis
The evolution in thinking about the pathogenesis of psoriasis. The inflammatory immune system has long been implicated in psoriatic disease.[66] In the past decades, tremendous advances have been made in understanding the pathogenesis of psoriasis.
When immunocompetent cells in exacerbating, untreated psoriatic skin were subjected to immunophenotyping using monoclonal antibodies in a two-stage immunoperoxidase technique, numerous epidermal changes were reported: accumulation of immunoglobulins in the stratum corneum, focal accumulation of OKM-1 positive but Mo-2 negative cells in the epidermis, reflecting granulocytes in Munro’s abscesses, decreased epidermal Langerhans cells, and sporadic exocytosis of mainly T-lymphocytes (T1, T8).[67] When quantified in patients with a variety of skin conditions, including but not limited to cutaneous psoriasis, epidermal infiltration of T-cells was most pronounced in psoriasis and least pronounced in atopic dermatitis.[67] In normal and diseased skin, the so-called “memory” T-lymphocytes (CD4+, CDw29+) were more numerous than “naïve” (virgin) T-cells (CD4+, CD45R+). A memory T-cell is one that has been activated by an antigen in the presence of antigen-presenting cells at some time after export from the thymus; this differentiation between a memory and a naïve cell also is associated with a change in the CD45 gene product. This predominance of memory cells over naïve cells suggests that most of the T-cells in diseased and normal skin are already primed and have met their specific ligand.[67]
In the early 1990s, it was thought that the primary pathogenesis of psoriasis involved keratinocytes and T-cells. A fusion protein made up of IL-2 and fragment of diphtheria toxin (DAB389IL-2) had been tested.[68] DAB389IL-2 selectively blocked activated lymphocytes (but not keratinocytes) and resulted in marked clinical improvement in psoriasis, with a concomitant reduction in intraepidermal CD3+ and CD8+ T-cell noted.[68] A T-cell model for psoriasis evolved at the time. In healthy skin, there was homeostasis with relatively low proliferation and complete differentiation. Activated CD25+ T-cells interacting with keratinocytes could trigger epidermal hyperplasia, leading to the characteristic high proliferation and incomplete differentiation observed in skin psoriasis along with a concomitant induction of immune-related surface proteins. The strength of this model was reinforced when it was observed that depletion of T-cells by DAB389IL-2 could clear psoriasis.
Thinking changed around 2003 when two biologic agents targeting T-cells (alefacept and ealizumab) were approved by the FDA. Both of these drugs were able to improve psoriasis and achieved scores of PASI 75 in more than half of all patients treated. However, there was no characterization of pathogenic T-cell subsets, associated cytokines, or cellular/molecular drivers of pathogenic immunity. It was then learned that CD4+ and CD8+ T-cells are activated by a dendritic antigen-presenting cell (APC) and an antigen. For example, CD8+ T-cells require that an antigen be presented by an APC known as MCH-1. MCH-1 favors intracellular antigens, such as viruses. Meanwhile, CD4+ T-cells are activated by MCH-2, which favors extracellular antigens, such as bacteria. Thus, psoriasis was once thought to be the result of dysregulation of the keratinocyte signaling system. This was later revised to a model that implicated autoimmune T-cell-mediated pathways. The introduction of new biologic agents, such as alefacept, ought to have confirmed the T-cell hypothesis, but instead alefacept was associated with a narrow therapeutic index, limited efficacy, and side effects. In retrospect, neither alefacept nor efalizumab were specific for any T-cell driven pathway. Instead, subsequent agents targeted TNFalpha and were more effective and better tolerated. Indeed, the introduction of TNFalpha blockade has revolutionized our current thinking about psoriasis and helped to clarify the crucial role of Th17 in psoriasis pathogenesis.[69]
Further elucidation indicates that there are multiple mechanisms that can activate dendritic cells, which initiate the psoriasis cascade. The dendritic cells can be induced to produce interferon-alpha by antimicrobial peptides (LL-37), which can complex with self-RNA and DNA to activate endosomal toll-like receptors (TLR) 7, 8, and 9.[70, 71] Localized in psoriatic lesional skin, plasmacytoid dendritic cells naturally produce IFN-a and might drive early Th1 cytokine expression before differentiating into tissue-resident dendritic cells or myeloid dendritic cells.[72] Resident in tissue cells, there may be a subpopulation of myeloid dendritic cells that express CD11c but not CD1c and produce inducible iNOS, IL-20, and IL-23. TNFalpha blockade inhibits IL-20 and IL-23,[73, 74, 75] and it is known that IL-23 levels correlate with psoriatic plaque development.[76] IL-6 and transforming growth factor beta1 upregulate the IL-23 in naïve T-cells, which allow some to differentiate into the Th17 subtype, so named because they produce IL-17.[77, 78]
Research suggests that Th17 cells are crucial to psoriasis pathogenesis. IL-17, a cytokine of the Th17 family, induces an inflammatory response that can be seen in psoriatic skin by the presence of neutrophils.[79] TNF-a acts with IL-17 in a synergistic fashion on keratinocytes, but IL-17 inhibition down-regulates these synergistic genes more extensively than TNFalpha inhibition.[80] This likewise points to the key role of Th17 in psoriasis,[81] with IL-17 contributing to the associated inflammation. It is likely IL-22 (secreted from Th17 and Th22 cells) induces the epidermal proliferation and de-differentiation of cells characteristic of psoriatic skin disease.[82–84] Furthermore, IL-22 up-regulates keratin-17, which has been associated with psoriasis, by way of JAK-STAT.[85] Today, psoriasis is one of the best understood autoimmune diseases.
Psoriasis features explained by Th17. A transcriptome may be considered a disease road map that shows the genes and pathways involved in a particular condition. From skin biopsies of patients with psoriasis, today we can identify 4,175 probe sets representing 2,725 unique, known genes associated with psoriasis, compared to only 159 in 2001.[86,87]
IL-22 is a cytokine produced by activated Th1 and other cells; IL-22 acts on epithelial cells and regulates the expression of only a few genes in keratinocytes.[88] These IL-22-regulated genes include psoriasin (S100A7), calgranulin A (S100A8), calgranulin B (S100A9), profilaggrin, keratin 1, keratin 10, kallikrein 7, matrix metalloproteinease (MMP) 1, MMP3, and desmocollin.[89] The effects of IL-22 are transcriptional and may be independent of protein synthesis and secretion or mediated via a secreted protein. Inflammation may enhance and amplify the effects of IL-22. Although classified as an interleukin, IL-22 does not appear to influence immune cells because IL-22R1 expression is evident in neither resting nor activated immune cells.[89] Patients with psoriasis exhibit elevated plasma levels of IL-22, which can be correlated with disease severity. Psoriasis therapy reduces the expression of IL-22 and IL-22-regulated genes. Cytokines of the IL-10 family (IL-19, IL-20, IL-22, IL-24, and IL-26) are up-regulated in psoriatic skin and may induce acanthosis in reconstituted human epidermis in a dose-dependent fashion.[90] Thus, IL-22 promotes acanthosis and impairs terminal differentiation.
By contrast, IL-17 up-regulates numerous genes in keratinocytes including innate defense molecules (DEFB4, S100A7, S100A12, S100A8, and S100A9), cytokines (IL-1F9, IL-8, IL-1Beta), and chemokines (CCL20, CXCL6, CXCL1, CXCL2, CXCL3, and CXCL5).[91] Together, IL-17 and IL-22 induce key molecular features of psoriasis. Keratinocytes have unique responses to IL-17 and IL-22, which are distinct from reactions to Th1 cytokines. In fact, Th1, Th17, and Th22 are the T-cells that drive cellular and molecular features of psoriasis by activating complex cytokine circuits.[92]
Ustekinumab is a human monoclonal antibody to the human IL-12 p40 subunit (thus, an anti-IL-12p40 agent), which was found to be effective in treatment of plaque psoriasis. In a study of 18 patients with psoriasis covering at least three percent of the BSA, 67 percent of the patients achieved at least 75-percent improvement in their PASI score over the course of the 16-week study.[93] Subsequent studies (PHOENIX 1 and 2) align with these results, where ustekinumab 45mg or 90mg produced PASI 75 at 12 weeks in 67 and 66 percent of patients, respectively (p<0.001 compared to placebo, 3%),[94] and in 67 and 76 percent of patients, respectively (p<0.001 compared to placebo 4%).[95] Histology likewise confirms the resolution of psoriasis with ustekinumab treatment.
The contributions of Th1 versus Th22 versus Th17 to psoriatic disease. Some clinical trials advance medical science by what they do not show. Humanized anti-IFN-gamma (fontolizumab) was studied for its effectiveness in the treatment of psoriasis. IFN-gamma is a pro-inflammatory cytokine that has been implicated in psoriasis and in other conditions, such as Crohn’s disease. However, the study ended in near total failure with 9 out of 10 of the patients treated with high doses of fontolizumab having no major improvement in psoriasis.96 This led to the conclusion that IFN-gamma (Th1 T-cells) may not be important in maintaining psoriasis.
This, in turn, led to a series of discoveries that pointed to the role of IL-17 in the psoriasis cellular/molecular phenotype in vivo. Direct antagonism of IL-17 began with IL-17 antibodies first introduced around 2009. IL-17 adapted for host-protective functions, but it appears that deranged IL-17 signaling may be associated with immunopathologies.[97] IL-17 blockade emerged as an important therapeutic target for psoriasis treatment as well as the treatment of other conditions, such as rheumatoid arthritis and Crohn’s disease. A short summary of the IL-17 family appears in Table 4.
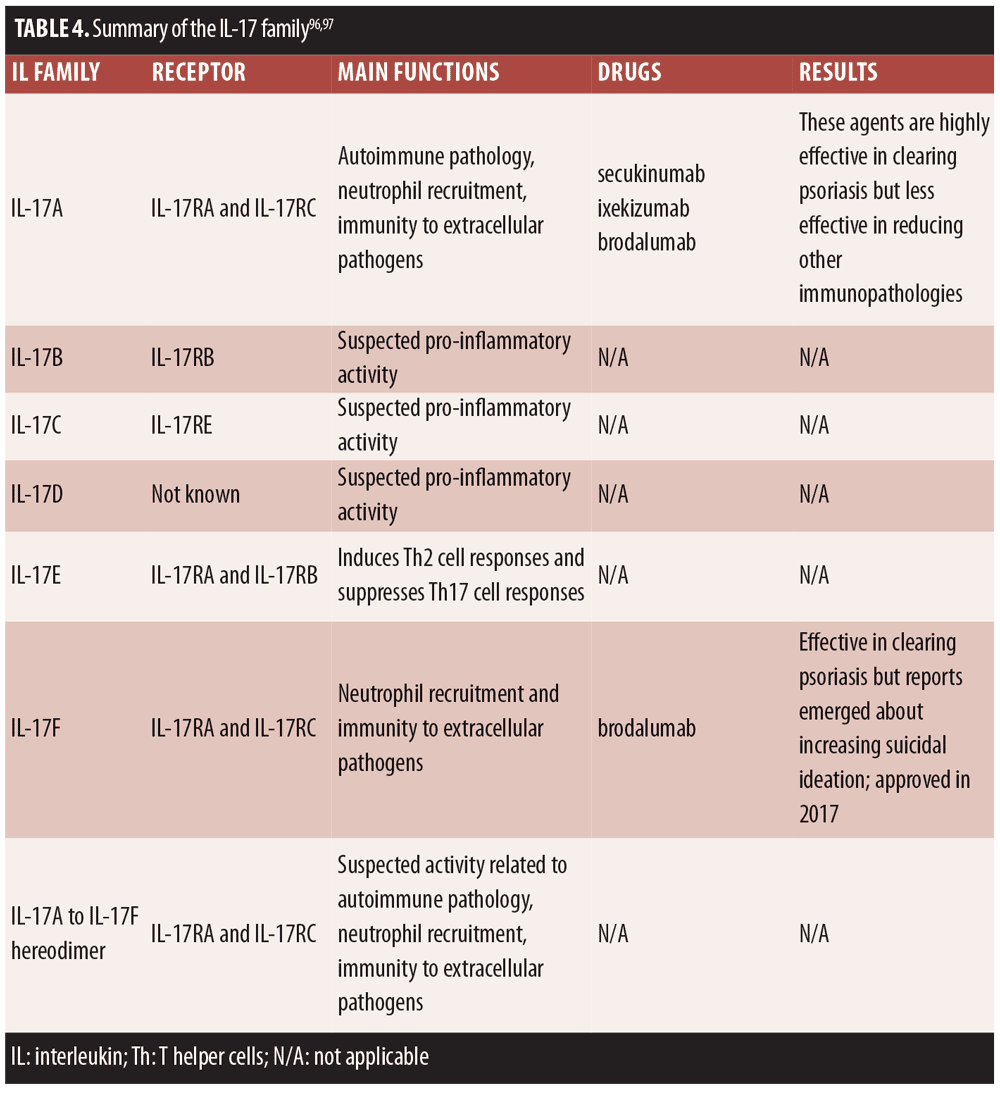
To date, six IL-17 family members have been identified, of which IL-17A and IL-17F are the best characterized. Increasing elucidation of the IL-17 cytokine family suggests there may be significant crosstalk among the various members.[97,98]
The first description of results obtained from a clinical trial evaluating IL-17 blockade was published by Hueber et al in 2010.[99] The drug, AIN457, was later market released as secukinumab. In this first clinical trial (n=104) of 12 weeks, the active agent significantly reduced psoriasis, sometimes as early as 2 to 4 weeks. The majority of adverse events were mild to moderate, no patients discontinued treatment because of lack of tolerability, and there were no deaths.[99]
In 2012, Papp et al conducted a randomized Phase 2 study that investigated the effects of AMG 827 (brodalumab) on 198 patients with plaque psoriasis with a minimum of 10% BSA involved.[100] IBrodalumab significantly reduced psoriasis in 12 weeks, with 72 percent of patients achieving PASI 90 scores. Adverse events occurred, the most common of which were nasopharyngitis, upper respiratory tract infection, and injection-site erythema; two cases of neutropenia were reported in the group that received brodalumab 210mg.100 Brodalumab was able to reverse gene expression across thousands of genes in psoriatic skin.[101]
Ixekizumab was evaluated by Leonardi et al in a 12-week Phase-2, placebo-controlled study of 142 patients with moderate-to-severe psoriasis. Significant improvement with ixekizumab versus placebo emerged in some patients in the first week.[102] Secukinumab, ixekizumab, and brodalumab were all able to target IL-17 and produce dramatic results in psoriatic skin, but the question remained as to the potential impact of maximal ligand blocking to suppress the entire downstream pathway. Upstream from Th-17 (and IL-17) is IL-23. It was hypothesized that IL-23 might control the entire TH17 pathway,[103] and the potential blockade of IL-23 and Th17 was proposed as a topic for further study. The humanized anti-IL-23 antibody guselkumab was evaluated in the treatment of moderate-to-severe psoriasis with promising results.[104] In a 28-week study, 70.0 percent of guselkumab patients versus 2.4 percent of placebo patients achieved PASI 90 scores at 16 weeks.[105] Risankizumab, a relatively new IL-23 antibody,[106] was shown to markedly reduce psoriasis in a Phase 2 clinical trial (n=166), providing significantly superior results to the active comparator in the trial, ustekinumab.[107] By the 12th week, the percentage of patients who achieved at least a 90-percent reduction in their baseline PASI score was 77 vs. 40 percent for risankizumab and ustekinumab, respectively (p<0.001). Serious adverse events were reported in all but one of the study arms: risankizumab 18mg (12%), risankizumab 90mg (15%), risankizumab 180mg (0), and ustekinumab 45mg or 90mg based on body weight (8%).[107]
Other IL-23 antagonists currently being studied include B1644066 and tildrakiuzumab. It appears that the IL-23/Th17 axis is crucial to sustaining psoriasis over time, and biologics that target this axis are showing dramatic results, which allow dermatologists to claim that psoriasis is the most effectively treated of all human autoimmune diseases.[108] This has led to the elucidation of the feed-forward IL-17 pathogenesis model where autoantigens directly or indirectly mediated by IL-17 trigger IL-23/IL-17 pathways, which begin a cascade including myeloid dendritic T-cell interactions; activated T-cell subpopulations; and the production of IL-17A, IL-22, and IFN-gamma, which in turn allow keratinocytes to “feed-forward” the inflammatory mediators and then amplify cellular immunity and sustain chronic T-cell activation.[108]
Thus, psoriasis vulgaris may be regarded as the chronic and maladaptive activation of the body’s natural immune response aimed at eliminating the Candida albicans and possibly other extracellular pathogens. However, psoriasis is driven by an autoantigen rather than an exogenous one. It has long been noted that patients with psoriasis are generally highly resistant to Staphylococcus aureus infection in contrast to patients with atopic dermatitis. Indeed, the pathogenic axes of certain cutaneous diseases are distinct and defined by a type of polar axis (Table 5).

Cellular and Molecular Profiling of Mild Psoriasis
Psoriasis occurs in all populations around the world, but frequencies vary. The disease is less prevalent in Asia than in the United States, and in the United States it is more common in Caucasians than African-Americans. There are also different subtypes of psoriasis.[109,110] Asian populations are more likely to develop small plaque psoriasis compared to severe psoriasis, which is more widespread in the West. In a study of 51 patients with biopsy-confirmed psoriasis, plaque size was measured (<2cm = small plaque) and disease severity assessed by PASI. Genomic and histologic studies were done to define psoriasis subtypes. A gene set variation analysis was conducted to determine which genes were expressed in the population. These results validated that Asian small and intermediate psoriasis were phenotypes and shared a common psoriasis transcriptome and histology with Western psoriasis (psoriasis vulgaris). Disease progression was then evaluated. Psoriasis spreads by vertical growth and radial expansion. However, Asian small plaque psoriasis revealed limited spreading but with great expression of proinflammatory cytokines regulated by IL-17A and IL-17.[109] In Western psoriasis, IL-17A- and IL-17-mediated cytokines were lower, but Western patients had many more T-cells and dendritic cells in their psoriatic skin areas. Both groups had diminished presence of negative immune regulators (CD69 and FAS), and both groups had incidences of arthritis and obesity, correlating with psoriasis severity. Based on this comparison, it seems that the dysregulation of T-cell expansion allows for the down-regulation of immune-negative regulators in large plaque psoriasis.[109]
Psoriasis is a T-cell mediated condition and autoimmune disorder, but the mechanisms by which autoantigens trigger T-cells in psoriasis continue to be elucidated.[111] Dendritic cells (DCs) stimulate autoimmune T-cells, and an antimicrobial peptide (AMP), specifically LL37, is present at high levels in psoriatic lesions and has been implicated in this process.112–115 A cationic peptide, LL37 may offer antimicrobial protection to the skin, and a recent study found that LL37 could be considered an autoantigen. LL37-specific T-cells produce pathogenic cytokines, including IL-17. This may be the first example of an AMP that stimulates both the body’s innate[112–114] and adaptive immune cells in the setting of autoimmune disease.[111] About two-thirds of patients with moderate-to-severe plaque psoriasis have CD4(+) and/or CD8(+) T-cells specific for LL37.[111]
The risk gene for psoriasis is HLA-C*0.6:02 on locus PSOR1 (6p21.33).115 It has been observed that epidermal CD8(+) cells must be present for skin psoriasis to develop.[116] Melanocytes have been identified as targets of the psoriatic T-cell response.[116] Melanocytes are the only epidermal cells that express ADAMTSL5, an ADAMTS-like protein that acts as a melanocytic autoantigen. ADAMTSl5 stimulation also induces cytokine, IL-17A, and CD8(+) T-cells, but only in patients with psoriasis, which suggests it is a psoriatic autoantigen.116 Thus, IL-17 directly or indirectly regulates autoantigens in psoriasis.
The emerging hypotheses are that there are Type 17 T-cell clones in psoriatic lesions, probably tissue-resident memory (Trm) phenotypes. At some point, tolerance to self-antigens would be broken. It is expected that regulating functions on T-cells in patients with moderate-to-severe psoriasis is partial at most. T-cells may potentially be restrained to some degree by the expression of negative immune regulatory pathways in a process known as “checkpoint inhibition.” In a collaborative project by Clark,117 psoriasis lesions were treated to resolution with etanercept. The lesions were then subjected to T-cell antigen receptor (TCR) sequencing, because it was thought that pathogenic Trm T-cells would remain in the skin at their previous lesional location. Vbeta-2, -13, and -16 clones were identified, which indicated that different clones shared the same CDR3 sequence, suggesting that they share reactivity to the same antigen.
Antigens, constitutive in normal skin, are up-regulated either directly or indirectly by IL-17 in keratinocytes or melanocytes. Trm T-cells may be reactive cells and will probably remain lifelong at their location and serve as memory for psoriasis antigens. CCL20, a substance induced by IL-17, triggers dendritic cell infiltration (both TIP-DCs and mature DCs) with potential for strong stimulation of T-cells in the skin. Note that CCL20 is also chemotactic for Th-17.
There are a number of negative immune regulators associated with psoriasis, including lymphocyte activation gene 3 (LAG3), cytotoxic T lymphocyte-associated 4 (CTLA4), indoleamine 2,3 dioxygenase (IDO1), apoptosis, programmed cell death 1 ligand (PDL1), programmed cell death 1 ligand 2 (PDL2), and IL-10. A study recently reported decreased expression of these in immunochemistry studies of psoriatic skin.[118] Thus, psoriasis chronicity may be related to the absence of negative immune regulatory pathways, and these negative pathways may be potential therapeutic targets. Furthermore, the researchers hypothesize that negative immune regulation limits disease progression in mild forms of psoriasis vulgaris and in the small-plaque Asian variant of psoriasis.
When psoriasis is described as “mild,” it typically refers to a reduced BSA affected rather than specific skin pathology. In general, mild psoriasis affects less than 10 percent of the body surface, accounts for about 80 percent of all psoriasis cases, and responds to topical treatment. Moderate-to-severe psoriasis affects over 10 percent of the BSA, accounts for 20 percent of psoriasis cases, and may require systemic therapy. However, even patients with mild psoriasis are still at greater risk for cardiovascular disease than normal controls. The comorbidities associated with severe psoriasis also occur in mild psoriasis. While systemic treatment is not always appropriate for mild psoriasis, a case-by-case evaluation may be warranted, weighing the potential risks (comorbidities) and benefits for the individual patient. The high expression of negative immune regulators creates the potential to modify disease if, for example, systemic treatments targeting IL-17A/F are employed.
This leaves an important and as-yet unanswered questions about psoriasis: If one could deplete or substantially reduce effector T-cell clones in mild plaques, would there be enough checkpoint inhibition or Treg function to prevent reactivation of disease-causing clones? Our current understanding of mild psoriasis includes some important points. First, it is not always “mild” in terms of the skin reactions to underlying T-cells. Topical products are the first-line approach to mild psoriasis, but their effectiveness can be limited particularly in comparison to systemic agents. Also, patients with mild psoriasis are at increased risk for comorbidities. Given these facts, we might ask if there is a subset of mild psoriasis patients who ought to be treated with relatively safe biologics or other systemic agents.
The answers to these questions are not yet clear but pose some important ideas that may potentially alter how we treat patients with mild psoriasis.
Cutaneous Oncology
From UV to SCC
Skin cancer remains by far the most common malignancy in humans in the world, including in Europe.[119] In terms of tumor etiology, nonmelanoma skin cancer (NMSC) should be included, as it encompasses basal cell carcinoma (BCC), Bowen’s disease, cutaneous squamous cell carcinoma (SCC), and its early antecedent, actinic keratosis (AK).[120] BCC is the most frequent form of invasive cancer around the world; in Germany, individuals approximately have a 30-percent lifetime risk of developing a BCC.[121] The risk factors for skin cancer are well known and include ultraviolet (UV) radiation, the human papillomavirus (HPV), and immunosuppression.[120] HPV has been implicated in benign and malignant cutaneous lesions and plays what seems to be an active role in the pathogenesis of certain cutaneous cancers.[120] Papillomaviruses are small DNA viruses found in more than 20 mammalian and avian species; the genome is composed of nearly 8,000 base pairs. HPV may infect keratinocytes of the skin and mucosa.[122]
The genital types of HPV are distinct from cutaneous HPV in that the virus does not integrate with the host genome in the skin. Integrated E6 and E7 genes can be observed in genital forms of the HPV. HPV23 and HPV38 may be a cofactor in certain cutaneous cancers, particularly in immunosuppressed patients, such as transplant patients, HIV patients, or patients with other autoimmune disorders under pharmacological immunosuppressive therapy.
While BCC typically presents as a single tumor, AK is best described as a field cancerization, which is described as a continuum in which precancerous and cancerous cells reside in an area of skin that may be adjacent or near skin that does not appear to be involved in the cancer.[123] Using the continuum paradigm, AK commences with photodamage and culminates in invasive SCC. Similar but differentially expressed genes found present in both AK and SCC suggests that AK may be a precursor lesion of SCC.[124] Gene expression has been studied in photodamaged and photoaged skin. Sun exposure can prompt the down-regulation of specific genes, which starts very early with photodamage and photoaging and precedes to more overt signs of AK over time. This raises two important questions: 1) Where should treatment begin? and 2) What is the role (and timing) of prevention?
The cascade of precipitating events that occurs in the etiology of AK occurs at different stages at multiple sites, such that photodamage, early AK, AK, and SCC may occur in close proximity in the same field. The paradigm in AK treatment has changed in recent years, such that it is now deemed important to treat the entire field rather than the visible lesions alone.[123] The classical pathway for AK development begins with photodamaged skin, which leads to a down-regulation of cutaneous immune response and continually expands the keratinocyte intraepidermal neoplasia (KIN). It had previously been accepted that this classical pathway involved several sequential steps, from AKI to AKII, AKIII, and finally SCC, but new evidence suggests that steps can be skipped, such that AKI can advance directly to SCC without the intervening stages.[125] In fact, in Germany, AK is accepted by clinicians as a form of carcinoma in situ.
The treatment algorithms for AK vary depending on whether solitary lesions or the entire field is treated (Figure 3).[127] Surgery remains the primary treatment for malignant melanoma and SCC, while noninvasive procedures are the first-line approach to AK.[126] Photodynamic therapy (PDT) as field therapy for AK is an effective therapeutic option, but its clinical utility is limited by associated pain. So painful is PDT that it averages 7.8 on an analog pain scale where 0 is no pain and 10 is the worst pain imaginable. As an alternative to conventional PDT, so-called “daylight PDT” has been developed.

Regardless of the algorithm, the goal of AK treatment is to address the AK present and prevent SCC in the field with fewer side effects, better safety, enhanced efficacy (low recurrence rate, prevention of development of invasive SCC), cost-effectiveness, and with a product that optimizes patient adherence (e.g., easy to apply in a short period of time). The presence of solitary AKs indicates that the patient’s skin is photodamaged and that he or she is at risk for developing skin cancer. Thus, a topical immune response modifier (IRM) may be appropriate for field treatment in that it treats existing lesions, subclinical AK, and may prevent AK from developing into SCC.
Next-generation imiquimod
New drugs are being developed for the safe, effective treatment of field cancerization, such as resiquimod, a next-generation imiquimod product aimed at treating AK.[128] Resiquimod is an imidazoquinoline that acts by cytokine secretion from monocytes and macrophages and has been shown to be effective in BCC and SCC in both immunocompetent and immunosuppressed patients.[129] Resiquimod is 100-fold more potent than imiquimod and can be used topically at 0.03% with a 90-percent complete clearance rate.[130] In fact, Szeimies et al[130] showed that more than 70 percent of patients achieved complete clearance after a single course of treatment. Resiquimod is applied topically for two or three days only. Adverse effects are the development of flu-like symptoms that may last up to 15 days; these were reported by 0 (0.01% gel), 3 percent (0.03% gel), 13 percent (0.06% gel), and 12 percent (0.1% gel) of patients.[130] Resiquimod may also be effective in the treatment of cutaneous lymphoma.
Topical ingenol mebutate gel may be used to treat AK. In one study, patients were treated for three days with ingenol mebutate gel 0.015% on the face or scalp or for two days on the trunk or extremities with 0.05% gel.[131] Of those patients who achieved complete clearance of the face or scalp lesions, most were enrolled in a 12-month study to evaluate recurrence rates. The sustained lesion reduction rates over baseline were 87.2 percent for face and scalp and 86.8 percent for trunk and extremities, with the estimated median time to recurrence set at 365 days (face/scalp) or 274 days (trunk/extremities).[131] No safety concerns were reported during the follow-up period of 12 months.
A pooled analysis of ingenol mebutate gel (0.015% for face/scalp AK and 0.05% for AK on the trunk or extremities) found ingenol mebutate was significantly more effective than placebo at clearing AK on the face or scalp (42.2% vs. 3.7%, p<0.001) as well as on the trunk or extremities (34.1% vs. 4.7%, <0.001).[132] Local skin responses peaked at about Day 4 and diminished to baseline by about Day 29.
Ingenol mebutate has a dual mechanism of action in the treatment of both melanoma and SCC. It acts on epidermal cells causing mitochondrial swelling and disruption of the cell membrane, culminating in apoptosis.[133, 134, 135, 136] This cascade leads to ingenol mebutate’s immunostimulatory effects and precipitation of the inflammatory response,[136] namely that it increases T-cell activity, increases dendritic cell activity, activates endothelial cells, including IL-1beta and TNF-a, releases cytokines[135] and recruits neutrophils.[134] Activated beta cells release tumor-specific antibodies that are then bound to antigen-bearing tumor cells; neutrophil binding to the Fc portion of tumor cell-bound antibodies results in the activation and release of reactive oxygen species which, in turn, leads to apoptotic cell death and avoids relapses.[134, 135, 137]
Low-dose 5-fluorouracil (5-FU) 0.5% combined with salicylic acid 10% can be used as a topical treatment for hyperkeratonic AK. In a proof-of-concept study (n=15, 66 AK), treatment occurred three times weekly for four weeks. At the end of 12 weeks, 77 percent of patients had complete clearance, 21 percent had partial clearance, and two percent were nonresponders.[138] When compared with diclofenac 3% in hyaluronic acid and vehicle,
5-FU 0.5% had greater histological and clinical clearance rates with durable benefits in the lesion-directed treatment of AK than the diclofenac with hyaluronic acid or vehicle.[139] In a placebo-controlled, double-blind, parallel-group, multicenter trial, AK patients were randomized to topical 5-FU 0.5% gel applied once daily, vehicle treatment, or diclofenac in hyaluronic acid twice daily.[140] Patients were treated for a maximum of 12 weeks, and lesion recurrence was evaluated at 6 and 12 months after the end of treatment. At 12 months, there was no recurrence of AK lesions in the 5-FU 0.5% gel group for 85.8 percent of patients compared to 79.8 percent of vehicle patients with no recurrence (p=0.04419) and 81.0 percent of patients in the diclofenac hyaluronic acid group (p=0.0246). Adverse events occurred most frequently in the 5-FU group (inflammation, burning) but generally did not cause patients to discontinue treatment.[140] In a Phase 3 randomized, double-blind, vehicle-controlled multicenter study (n=166), patients with field AK were randomized to receive either 5-FU 0.5% gel combined with salicylic acid 10% or vehicle applied topically once a day for 12 weeks (contiguous areas of the skin for 25cm2).[141] The first and last topical applications were performed by the study clinicians but otherwise patients applied the medication themselves. After eight weeks of treatments, the intention-to-treat and per-protocol populations of this study had significantly better results with 5-FU 0.5% gel and salicylic acid than the vehicle, 49.5 vs. 18.2 percent for intention-to-treat patients (odds ratio 3.9, 95% CI: 1.7–8.7, p=0.0006). Patients in the active-treatment group had more application and administration site reactions compared to vehicle patients (99.1% vs. 83.6%). The study results of both groups remained similar and trended in the same manner during the 12 weeks of treatment but diverged markedly in the eight weeks after treatment stopped (Figure 4).

Cryotherapy is appropriate for single, well-defined, hyperkeratonic AK lesions. Field treatment represents a paradigm shift but treats AK as carcinoma in situ and recognizes that there are subclinical lesions in the field that require treatment as well. Topical agents are particularly appropriate for field therapy in that they can treat broader areas and address the subclinical lesions as well. Increasingly, these two therapies are combined for optimal patient management.
Ruminations on the War on Cancer
The language physicians use to talk about cancer has always been the language of war: patients fought battles, researchers and their sponsors declared war, targets were identified, tumors invaded, the latest drugs and treatments were put into our clinical armamentarium, and everyone wanted to find that elusive “silver bullet.” A more realistic assessment today is that we understand more about cancer’s pathogenesis and the genetic underpinnings of the disease than ever, but we remain unable to consistently cure many types of cancer. Targeted therapies in which so much promise resided are not always effective and results are often not robust. Cancer is adaptive and elusive, and it may be time to drop the war-time metaphors.[142] For example, rather than talking about cutaneous malignancies as cancer or precancer, perhaps it is better to talk about “indolent lesions of epithelial origin” and to recognize that this disease is a continuum of events that can be treated at any number of points.
The new epidemic of BCC. Recent studies from around the world are calling the observed increase in BCC an epidemic.[143,144] In northern California, the rate of BCC increased about 13 percent from 1998 to 2012. The exact incidence of BCC is difficult to assess, because BCCs are not reportable cancers and do not have unique ICD-9 identifiers. The rate of BCC in the United States is estimated to be two million.145 In the clinic, BCC risk stratification can be based on two broad categories: clinical factors and histology. Clinical features include the tumor’s size and location, the depth of invasion, and whether the tumor had recurred, gone through-and-through, or had previously undergone radiation therapy (Table 6).

SCC. SCC occurs less frequently than BCC but it is likewise increasing, possibly due to better detection efforts, more exposure to sunlight, and increased longevity. About 2,000 Americans die each year from some form of skin cancer.[146] An elevated risk exists in patients with SCC with tumor diameter more than 2cm, Breslow depth more than 2mm, ulceration, a site of the SCC on the ear or non-hairy lip, poor differentiation (grade 3+), and perineural invasion. SCC may be treated with oral capecitabine, a prodrug that converts in the body to 5-FU.147 Capecitabine is effective in treating forms of colorectal cancer, breast cancer, and gastric cancer, but it may be effective in treating SCC as well.[148]
The role of PDT in cutaneous oncology. The role of PDT in treating skin cancers, such as superficial BCC, nodular BCC, and SCC, is a subject of growing importance. Early studies found PDT could achieve good clearance results for many forms of cancer, perhaps on par with radiation, but recurrence rates were higher than with surgical excision or Mohs micrographic surgery (MMS). The use of multiple PDT sessions and the pretreatment with penetration enhancers, such as 5-aminolevulinic acid (ALA), are providing promising new data. Certainly, PDT has been and remains an important treatment option for those patients with BCC and SCC for whom surgery must be precluded. Marmur et al[149] conducted a literature review of the use of PDT for treatment of skin cancer, finding encouraging results for some types of cancers (Table 7). The role of PDT in the treatment of invasive SCC is limited owing to potential metastases.

Bowen’s disease, an SCC in situ, may be considered an early stage or intra-epidermal form of SCC. Transplant patients have an elevated risk for cutaneous epithelial neoplasms, including AK and Bowen’s disease. A controlled trial (n=40) found that for transplant patients with skin cancer, PDT preceded by application of 20% ALA was a safe and effective treatment with good cosmetic results.[150] ALA-PDT may be particularly helpful when treating immunosuppressed patients with large regions of lesions.
Nevoid basal cell carcinoma syndrome (NBCCS) is an autosomal dominant disorder that is associated with congenital abnormalities as well as increased risk of medulloblastoma and early development of BCC.[151,152] In a single-center case series, three children with BCCs and basaloid follicular hematomas (BFHs) over 12 to 25 percent of their BSAs were treated with 20% ALA over up to 22 percent of their BSAs for 24 hours with occlusion.[153] They were then treated with PDT in fields up to 7cm (dye laser) or 16mm (lamp) with as many as 36 fields treated in one session. After 4 to 7 sessions, the patients had 85- to 98-percent overall clearance and excellent cosmetic outcomes with no scarring. The patients tolerated the treatments well, morbidity was minimal, and they healed quickly. Results were durable up to six years.[153] ALA selectively accumulates in the tumor rather than in normal skin.[154] Note that it may be necessary to use local anesthetic with ALA-PDT.
BF-200 10% ALA gel relies on a nanoemulsion (BF-200) to enhance transport of ALA through the stratum corneum down to the basal membrane, allowing the light source to achieve an even greater depth of penetration. The red light source recommended for PDT is the BF-RhodoLED. In a study comparing BF-200 10% ALA nanoemulsion gel to 20% ALA cream in 20 patients, BF-200 ALA was associated with more intense protoporphyin X (PpIX) fluorescence under fluorescence microscopy compared to 20% ALA cream.[155]
In a Phase 3 study from the European Union,[156] PDT with methyl aminolevulinate (MAL) was compared to BF-200 10% ALA gel for the treatment of BCC. A major statistical endpoint of this study was noninferiority of BF-200-ALA versus MAL, which was met. Complete clearance rates for patients (93.4% vs. 91% for BF-200-ALA and MAL, respectively) and complete clearance of all types of lesions (94.6% vs. 92.9% for BF-200-ALA and MAL, respectively) were similar. Thicker and more nodular BCCs showed better response to BF-200-ALA than MAL (Figure 5). Cosmetic outcomes were excellent for the BF-200 ALA patients. Thus, it appears that BF-200 ALA is very effective in the treatment of non-aggressive BCCs, particularly lesions less than 1mm thick, and it is superior to MAL for the treatment of nodular BCCs, possibly because of its deeper skin penetration.

Topical imiquimod 5% cream. Multiple case reports support the use of imiquimod 5% cream for the treatment of SCC in situ. In a case study of a kidney transplant patient with a history of metastatic prostate cancer, an invasive SCC in situ was treated at the hairline with 5% imiquimod cream applied three times per week and left on overnight (8 hours). After three weeks, the tumor had regressed noticeably but central erythema was still evident. Other than scaling, no adverse events occurred over the 12-week course.[157] Topical 5% imiquimod cream was used to treat a large facial SCC in situ (Bowen’s disease) in a 75-year-old woman with a history of Bowen’s disease. The cream was applied every other night over six weeks, resulting in total clearance at the end of the course and no recurrence observed in eight months.[158]
Combination therapy may be of value in particularly difficult cases, such as SCC on fingers where surgical intervention may compromise function. Four patients with SCC in situ on a digit were first treated with imiquimod cream as a monotherapy; all failed. Two of those patients also underwent 5-FU cream treatment monotherapy and failed.[159] All four patients were then administered imiquimod and 5-FU combination therapy for eight weeks with good response and no recurrence at 12 to 23 months after treatment.
In a retrospective analysis of histological biopsy specimens of superficial BCC, 127 cases were reviewed where the patient had been treated primarily with imiquimod five times per week over a six-week course with a mean follow-up period of 34 months (range 3–91).[160] Recurrence was clinically ascertained with histological confirmation. Among the nonrecurrent cases, median tumor thickness was 0.26mm (range 0.09–0.61), but recurrent cases had a significantly greater median tumor thickness of 0.57mm (range 0.41–1.41, p<0.0001). Lesions greater than 40mm had a recurrence rate of 58 percent, but there were no cases of recurrence for lesions 0.40mm or less. Thus, topical imiquimod may be an effective treatment option for superficial BCC.
A systematic review reported clearance rates for topical imiquimod 5% cream to be 43 to 100 percent for superficial BCC, 42 to 100 percent for nodular BCC, 56 to 63 percent for infiltrative BCC, 73 to 88 percent for SCC in situ, and 71 percent for invasive SCC.[161] It was noted that the more intense inflammatory reactions to treatment could be associated with higher rates of tumor clearance. The relative advantages and disadvantages of topical imiquimod 5% cream are briefly summarized in Table 8.

Ingenol mebutate gel. A retrospective chart review (n=7) of patients with superficial BCC treated with ingenol mebutate over 10 to 14 days following a shave biopsy was conducted.[162] Patients were treated for four weeks. All lesions were resolved by the end of treatment, and six lesion sites in four patients at 3 or 4 months post treatment were biopsied to confirm histological clearance. All patients experienced localized adverse events that began on the first day or two of treatment, peaked at two weeks, and then largely resolved at two weeks.
Intralesional chemotherapy. Intralesional chemotherapy can be used to treat BCC and SCC, but this approach is not mentioned in most guidelines, is not frequently employed, and has not been widely studied particularly with respect to long-term results.[163] A number of intralesional chemotherapeutic agents may be used, but there is little evidence in the literature to recommend one particular course over another (agent, dosing, frequency, and appropriate lab monitoring). IFN, 5-FU, and IFNa are among the chemotherapeutic agents appropriate for intralesional therapy. In addition, methotrexate is gaining recognition for its intralesional or perilesional use in treating solitary keratoacanthoma (KA).164 For smaller KA (<1 cm), a single injection from a peripheral site with the needle tip at the deeper central portion of the lesion should suffice; a larger KA may require multiple injections around the edges, all directed toward the center. In many ways, this technique is similar to injecting corticosteroids into hypertrophic scars. Local anesthesia may be needed prior to intralesional or perilesional therapy, particularly if the procedure is difficult or the patient is very anxious. Prior to treatment, it may be helpful to discuss treatment and listen to the patient’s objectives to make sure the patient does not harbor unrealistic expectations. It may be appropriate to use Luer-Lok™ syringes for the injection (30-gauge needle or similar). The clinician should consider eye protection and other protective measures when administering the injections. During injection, some of the chemotherapeutic agent may leak through the central crust or incisions from a previous biopsy; this should not diminish clinical efficacy. The patient should be advised that crusting and necrosis is normal and expected about 7 to 10 days after each injection. A gentle debridement of necrotic tissue may be useful prior to the next injection to make sure the injection is able to deliver the chemotherapeutic agent to viable neoplastic tissue. Injections should be carried out once every week or two; if the patient does not respond after two injections, a change in treatment should be evaluated. Some patients, such as those with renal failure, may require lab monitoring throughout the course of treatment. In some instances, a post-treatment biopsy should be considered as well.
The end to our war on skin cancer? There are many ways to manage skin cancers, and war metaphors may be outmoded. Topical and pharmacological approaches are less invasive and may be effective in many patients. Indeed, they may be the only option for fragile or compromised patients. These medical treatments are associated with less pain, improved cosmesis, and are effective in treating multiple lesions and subclinical disease. Avoiding surgery offers many benefits: it may save time, money, and patient anxiety and apprehension. Topical treatments have some drawbacks, however: no margin control and not appropriate for high-risk BCC or most invasive forms of SCC. Furthermore, there is a lack of long-term data on recurrence for the newer treatments. Thus, surgery must remain the “gold standard,” but today there are many new options to consider as well.
What’s New in Lentigo Maligna (Basal Cell) and Merkel Cell Carcinoma?
Confocal microscopy. Confocal microscopy is a technique that increases the optical resolution of an image by means of a spatial pinhole at the confocal plane of the lens; this eliminates out-of-focus light. Modern fluorescence-correlation microscopy (FCM) devices allow dermatologists to gain a quasi-histological view of a skin tumor. In fluorescence mode, FCM can be used to assess freshly excised specimens; in reflectance mode, tumor margins in vivo on the patient’s own skin can be assessed. Reflectance confocal microscopy (RCM) has been used bedside as a supplemental tool for mapping and monitoring of lentigo maligna (LM) and BCC.[165]
When evaluating BCC, cancer margins can be first marked out with dermoscopy and then re-checked with RCM. RCM showed BCC outside of presurgical mark in 30 percent of lesions.[165] This occurs because deep tumor margins cannot always be assessed with limited depth-laser penetration. RCM may also be an important tool for the rapid detection of residual tumor in a surgical wound. This may require a smaller microscope head with an automated approach so that the entire wound can be imaged in a rapid, controlled manner.
LM is a subtype of melanoma in situ, characterized by a lentiginous growth pattern of melanocytes as solitary units at the dermal-epidermal junction (DEJ) of chronically photodamaged skin. LM may have ill-defined borders and possible (and significant) subclinical extension. It can be challenging to preoperatively delineate LM because it occurs against the background of sun-damaged skin and there can be intrinsic, early changes in the tumor, which is composed of a single atypical melanocytic proliferation.[165] In a study of 29 patients with LM, margins were first obtained clinically using dermoscopic examination, and then RCM was used to obtain margins in no more than four radial directions per patient. In this study population, there were 4 out of 29 false positives where RCM-diagnosed LM could not be confirmed histologically. There were also 5 out of 29 false negatives, where LM could be confirmed histologically but was not evident with RCM.[165] RCM may also be used to monitor how LM responds to nonsurgical treatments, such as imiquimod. In a study, RCM could identify 70 percent of all imiquimod responders with no false negatives, which puts it roughly on par with histopathology.[165]
For freshly excised tumors, ex-vivo FCM can be used with different fluorophores at varying wavelengths (the most commonly used is acridine orange, which provides excellent contrast). For BCC, FCM correlates well with frozens, offering 88-percent sensitivity and 99-percent specificity. It may even offer more information in that FCM can help analyze fat tissues and other structures that could be altered in frozen Mohs processing. The fundamental limitations of ex-vivo FCM is that it can be challenging to recognize the cords and strands of infiltrative BCC and distinguish them from stroma. Sebaceous glands may be difficult to identify and may be confused with basaloid islands.[165]
FCM technology continues to evolve and improve, with the potential for a wide range of applications for dermatologists. Image databases could one day be set up and connected to artificial intelligence systems to allow for real-time diagnosis and to help guide treatments.
Melanoma. Subclinical spread describes the microscopic tumor extension beyond the margin, which can result in positive margins with wide local excision and may be associated with local recurrence, both of which complicate melanoma management.[166] MMS and related techniques can be used for more rigorous subclinical tumor identification for certain types of melanoma, but the clinical factors associated with the subclinical spread of in-situ melanoma remain to be elucidated. That frequency of subclinical spread of melanoma in situ occurs in 12 to 71 percent of cases.[166] In a study of 674 large melanomas,[166] the visible melanoma plus margins (2–3mm of normal skin) were removed with a debulking excision and sent for formalin-fixed, paraffin-embedded, broad loaf sectioning for staging. A Mohs layer was then taken with a 2 to 3mm margin for frozen hematoxylin and eosin (H&E) stain and MART-1 immunostain. If there were positive margins, additional stages were required. All tumors in this study had margins of at least 5mm. The study was limited by the fact that it was carried out at a single academic center, histological subtypes were not noted, and it did not account for all risk factors (e.g., photoaging degree, percentage of clinically visible tumor after biopsy, tumor color). This study concluded that the subclinical spread of melanoma in situ could be associated with the tumor location (highest risk were tumors on the head, neck, acral sites, and the pretibial leg), recurrence after previous treatment, preoperative size (>1cm), and increasing age (> 60 years, risk increases by about 2% each year). Risk increases with multiple risk factors. When clinicians triage surgical treatment of melanoma in situ, it is important to look at risk factors when choosing between standard excision and more exhaustive microscopic margin assessment.[166]
While invasive melanoma may require more rigorous margin assessment in order to remove subclinical tumor prior to reconstruction, the indications for these techniques have not yet been objectively defined. In a retrospective, cross-sectional analysis of 216 invasive melanoma, MMS with frozen-section, bread-loaf processing of the debulking excision for frozen H&E stain and MART-1 immunostain was taken.[167] All melanomas had subclinical spreading necessitating at least two stages of MMS to clear margins. For tumors classified T1a, a minimum 5 to 6mm margin was excised, while for T1b and above, a minimum of 1cm was excised. If the biopsy fulfilled the criteria for a sentinel lymph node biopsy, the patient underwent that procedure prior to MMS surgery. In the event that the tumor upstaged during frozen section evaluation of the residual tumor, patients were offered a sentinel lymph node biopsy before reconstruction. After MMS, a debulking excision was sent for paraffin-embedded sectioning to confirm staging and to archive the primary tumor. In this study, 83 out of 216 melanomas had evidence of subclinical spread. The clinical predictors for subclinical spread were tumor location (head and neck were highest risk), recurrence after previous treatment, size more than 1cm, and patient age of 65 or older. One histological predictor was determined, namely the presence of mitoses. Thus, it appears that there are comparable indications for MMS with rigorous microscopic margin assessment prior to reconstruction that might apply to both invasive and in-situ melanoma.
The local recurrence rates of head and neck melanomas were evaluated in an observational cohort study conducted at a single academic center from 1997 to 2006 with a median follow up of 9.3 years.[168] All patients (n=806) underwent staged excision to treat poorly defined melanomas with unpredictable occult extension. Local recurrence rates were 1.4, 1.8, and 2.2 percent at 5, 7.5, and 10 years, respectively. This study evaluated the association between the size of the lesion and the distance from the lesion needed to obtain a pathologically tumor-free margin. Invasive melanoma required greater margins than melanoma in situ. The mean margin from lesion to clearance for melanoma in situ was 9.3±5.1mm and was 13.7±5.9mm for invasive melanoma. Larger lesions recurred more often; for every 50mm2 increase in the size of the clinical lesions, the patient had a nine-percent increase in the rate of local recurrence. Local recurrence could also be associated the cutaneous tumor site, and immunosuppression (no immunosuppressed patient experienced a recurrence). This suggests that staged excision with comprehensive permanent section margin control of melanoma in chronically photodamaged skin on the head and neck confers on patients favorable recurrence rates when the melanoma margins are difficult to assess; recurrence rates are higher with traditional techniques. Recurrence rates may increase over the long term; in this study, about a third of recurrences (36%) occurred after five years. The larger lesion size was associated with the necessity for a greater margin for clearance, as was in-situ disease compared to invasive disease.[168]
A novel technique for achieving optimal sections during staged LM excisions has recently been described in the literature.[169] This approach was developed to help overcome the high recurrence rates of LM with convention excision procedures and conventional MMS. Since LM may be characterized by a wide subclinical spread, the traditional bread-loaf sectioning is unsatisfactory because it does not allow for complete visualization of margins. MMS relies on frozen tissue sections, which is not ideal for visualizing melanocytes. Some new melanocyte stains have been introduced that enhance visibility but they are associated with higher costs and greater effort.[170] A newer technique relies on formaldehyde-fixed, paraffin-embedded staged excision, which may permit better visualization. In this new approach, the apparent margins of the lesion are first delineated under dermatoscopic evaluation.[169]
When dealing with small lesions, two tissue strips (right and left) are excised vertically down to the deep fat layer; the 12 o’clock edge is marked in red, the six o’clock in blue. The outer edge of each strip is also marked in blue as this is the side embedded en face in the paraffin. At the time of the first layer, the central portion of the lesion is also excised and sent to the laboratory for standard serial sectioning to determine the possible presence of invasive disease. Thin strips of tissue are prone to warping in formalin, so this technique uses glass slides to keep tissue strips as flat as possible during fixation. This technique allows for high-quality en face paraffin-embedded sections, which enhance visualization of the margins. This type of staged marginal excision with mapping has emerged as an optimal approach for managing LM and LM melanoma.[161]
LM melanoma and malignant melanoma in situ are often treated with MMS with frozen section immunochemistry. Immunostains used in this connection have specific advantages and disadvantages. Melan-A works well on frozen sections, but may not offer sufficient specificity. Microphthalmia transcription factor (MITF) is a more specific nuclear melanocyte immunostain, but it is less frequently used in clinical practice. In a study (n=16) of patients with either malignant melanoma in situ or LM melanoma, frozen sections from chronic sun-damaged skin with negative margin and 12 tumor samples were stained with both Melan-A and MITF.[171] The mean melanocyte counts differed significantly (p<0.001) at 9.8 (MITF) and 13.7 (melan-A). The negative margins for the mean melanocyte counts likewise differed significantly (p<0.001) with values of 8.84 (MITF) and 14.06 (melan-A). The tumor mean melanocyte counts were 63.5 and 62.4 for MITF and melan-A, respectively. Thus, it appears that melanocyte density on tumor-free chronic sun-damaged skin is higher with melan-A than MITF although MITF provides a clear outline of the melanocyte nuclei.[171] It should be noted that there may be significant nonmelanocyte epidermal staining by melan-A in the negative margins and in chronic sun-damaged skin, but MITF is an effective alternative to melan-A that enhances nuclear size and pleomorphism and allows for accurate quantification of melanocytes. In terms of cost, stain time, and tissue processing, MITF and melan-A approaches are similar.
Merkel cell carcinoma. Merkel cell carcinoma is an aggressive skin cancer associated with UV light exposure and the Merkel-cell polyomavirus (MCPyV).[172] Advanced Merkel cell carcinoma may respond transiently to chemotherapy but median progression-free survival is poor at about three months.[173] Chemotherapy seems to confer little to no survival benefit with median survival around nine months; long-term survival is rare.[174] The programmed death 1 (PD1) immune inhibitory pathway appears to mediate localized immune resistance. PD1 comprises the PD1 T-cell coinhibitory receptors and ligands PD-L1 and PDL2, which are expressed in tumor and immune cells. MCPyV-specific T-cells further express PD1. A treatment strategy of “block-the-blocker” has evolved using pembrolizumab (humanized monoclonal IgG4 antibody that blocks PD1) or avelumab (blocks human monoclonal IgG1 that blocks PD1).[172]
Pembrolizumab was evaluated in a Phase 2 study of 25 patients with advanced Merkel cell carcinoma. Investigators found the objective response rate during treatment was 56 percent (95% CI: 35–76), with 4 out of 25 patients achieving complete and 10 out of 25 partial response.[172] Most patients in this study responded by Week 12. Adverse events were reported in 77 percent of patients, with 15 percent reporting more severe (Grades 3 or 4) events.
In 2017, the FDA approved avelumab, the first approved treatment specifically for Merkel cell carcinoma. In a study of 88 patients with advanced Merkel cell carcinoma previously treated with chemotherapy, nine percent of the avelumab patients achieved complete response and 23 percent achieved partial response.[175] Duration of response was at least six months for the majority of patients (86%). Thus, growing evidence suggests that checkpoint blockade should be considered the best treatment for achieving good response rates and durable results in patients with advanced Merkel cell carcinoma. Treatment-related adverse events occurred in 71 percent of patients, but most were mild. Avelumab represents an emerging standard of care for advanced Merkel cell carcinoma that shows promise.
First-line therapy with pembrolizumab offers an objective response rate of 56 percent, far surpassing conventional chemotherapy, and these results were observed in both MCPyV-positive and -negative tumors.[172] For that reason, PD-L1 expression should not guide clinical decision making in terms of whether to treat a patient with Merkel cell carcinoma with PD1 blockade. Immune checkpoint blockers should be considered and may be enhanced by the potential synergistic effect between immunotherapy and radiotherapy.[176]
PDT + Imiquimod + 5-FU: An Update
Evolution of PDT: The quest for short incubation and painless protocols. PDT is a well-established and familiar dermatologic treatment. It requires three fundamental things: oxygen (to create reactive oxygen species or ROS), a photosensitizer (PplX), and some sort of activating light source. Among the FDA-approved photosensitizers used are 5-ALA and methyl ester ALA, the latter of which is not marketed in the United States. The activating light source may be red or blue light. The 20% ALA with 14-to-18-hour incubation periods used in PDT today evolved from clinical trials and fluorokinetic analyses. These treatments could be effective but they involved protracted incubation periods and were painful for patients. In a recent vehicle-controlled study, ALA or vehicle for the treatment of AK was applied in broad-area application of face or scalp for one, two, or three hours or as spot application with two hours of incubation; following incubation, patients underwent blue-light activation.177 At eight weeks, median AK clearance rates following a single treatment ranged from 36 to 57 percent. At 12 weeks, the median AK clearance rates of 68 to 79 percent (ALA) were significantly better than the vehicle group (7%, p<0.0001). At Week 12, complete clearance was reported in 17 to 30 percent of ALA patients versus two percent of vehicle patients (p=0.0041).
With the goal of reducing treatment to a single PDT session, pretreatment can be used that may involve either 5-FU or imiquimod. Patients should apply the topical to the face (7 days) or scalp, trunk, or extremities (10 days). Imiquimod 3.75% or 5% is applied over the course of seven days on the face and scalp. Imiquimod should not be used for lesions on the trunk as there is a possibility of scarring. Another way to enhance PDT efficacy is microneedling, which potentiates penetration of the ALA or MAL. It is important to note that deeper microneedling is not approved by the FDA.
Increasing the temperature to 38.8° C during incubation of the extremities may also be helpful.[178] In a one-year study, 18 patients with at least 10 AKs on the upper and lower extremities (i.e., 20 pairs of extremities) were enrolled for ALA-PDT at a single center.[179] Following a one-hour incubation period, patients were exposed to 10 J/cm 417-nm blue light. The median baseline temperature of the treated lesions was 31.6° C, and the median maximum temperature during incubation was 41.2° C. Lesions were counted and photographed at baseline and one week, then three, six, nine, and 12 months after treatment. At baseline, there was a total of 724 Grade 1 or 2 AKs (median 15 on each extremity). At three months, the median clearance was 90 percent, with results maintained over 12 months. At three and 12 months, the lesion count was 70 and 72, respectively. Grade 3 lesions did not resolve with treatment. Warming the extremities after ALA application was well tolerated by patients and did not increase or exacerbate side effects.
New consensus guidelines for PDT have recently been published by the American Society of Dermatologic Surgery (ASDS) that recommend PDT as effective in the treatment of precancerous lesions, superficial nonmelanoma skin cancers, inflammatory acne vulgaris, and certain other conditions.[180] An important and potential treatment-limiting consideration in PDT is that more than 60 percent of PDT patients experience moderate-to-severe burning pain, and many patients report intolerable pain. In a prospective, split-face study comparing short ALA incubation times (15 minutes) with 60 minutes of blue light (“painless therapy”) versus conventional ALA-PDT, three patients underwent AK procedures in a single center.[181] The painless patients showed a 52-percent reduction in lesions compared to 44 percent for the conventional group. Patients rated pain during treatment on a scale from 0 to 10 (where 0 was no pain and 10 the most severe pain imaginable). Painless therapy was scored as 0 for pain at all time points, whereas the average pain score for ALA-PDT was 7. Thus, painless PDT is not only possible, it appears to be safe and effective.
BF-200 for PDT: new kid on the block. BF-200 10% ALA gel is a novel product for PDT and allows for deep PpIX induction down to the basal membrane. When this gel is used for PDT, the protocol should call for lesions to be lightly abraded before the gel is applied topically and then should be allowed to dry for about 10 minutes. A light-tight occlusive dressing should then be placed over the treatment area. A three-hour incubation period is recommended. At the conclusion of the incubation period, the dressing should be removed and any gel still on the surface should be wiped away. Three Phase 3 pivotal trials for AK patients were conducted in Germany comparing BF-200 10% ALA gel (Ameluz®, Biofrontera Inc., Boston, Massachusetts) to Metvix®/Metvixia® (Galderma Laboratories, Fort Worth, Texas), and placebo.[182,183] In all three studies combined, 779 patients with 4 to 8 mild-to-moderate AKs were enrolled. Complete clearance of all lesions after one or two PDT sessions was the primary endpoint. Complete clearance was achieved by 62 percent of BF-200 patients after one treatment and 91 percent achieved complete clearance after one or two sessions. In the comparator study between BF-200 PDT and MAL-PDT, BF-200 PDT was found to be non-inferior to MAL PDT. Most of the BF-200 patients experienced adverse events (96%) but the rates were higher with narrow-spectrum PDT compared to broad-spectrum PDT. Some of the most frequently reported adverse events were erythema, burning, and pain.[183]
Does PDT decrease the incidence of NMSC? The role of PDT with ALA 20% in reducing the rate of NMSC continues to be investigated. A 52-week study of patients at high-risk for AK compared ALA-PDT to placebo following cryotherapy and evaluated the development of NMSC.[184] The study included 166 patients with clinically evident facial AKs, a history of nonmelanoma skin cancer, and histological evidence of dysplasia within clinically normal-looking perilesional skin. Patients were first treated with cryotherapy and then randomized to one of three groups: ALA-2X (two ALA-PDT treatments, baseline, Week 4), ALA-3X (three ALA-PDT treatments, baseline, Week 4, Week 24), and VEH-PDT (placebo treatments that were matched in a 1:1 ratio to the two active groups). ALA or vehicle was applied to the full facial region an hour before blue light treatment, which lasted 16 minutes and 40 seconds. All patients were evaluated 24 hours after each PDT session. Significantly more ALA patients had zero AKs on the face at 52 weeks compared to the placebo (vehicle) patients.[175] In the safety population, it was determined that ALA-3X significantly decreased the rate of nonmelanoma skin cancer development over time (5 versus 12) compared to vehicle.
The FDA has approved numerous treatments for topical field therapy for AK, giving clinicians and their patients many options (Table 9). The burden of AK may further be reduced with liquid nitrogen cryotherapeutic treatments (1 to 4 times a year, or more, as appropriate), more field therapies, sunscreen education and recommendations, topical retinoid therapy (tretinoin or tazarotene), and vitamin B3. An alternative approach to reducing the AK burden may be a protocol involving a daily application of 3.75% imiquimod for seven days, followed by a two-week rest period (no treatment), then a once-a-week application of 3.75% imiquimod from then on (indefinitely, therapy may persist over years). This is an off-label therapy and should not be recommended for patients with autoimmune disorders or in immunocompromised patients.

Old school versus new school 5-fluorouracil. The old-school “scorched earth” approach using 5-FU was associated with burning pain, which could limit treatment. Yet, 5-FU was highly effective; in two Phase 3 vehicle-controlled trials, over 70-percent clearance was achieved with one single week of treatment.[185, 186] A new-school approach handles this in two cycles. In the first cycle, 5-FU is applied daily over seven days (face) or over 10 days (nonfacial areas). Patients are then given a one-month rest period when no 5-FU is applied. The second cycle calls for daily application for two or three more weeks (individualized for patient).
Disruptive Technologies for Skin Cancer: Electronic Brachytherapy and Superficial Radiation
Electronic brachytherapy (EBX) and superficial radiation offer the potential for long-lasting effective treatments of NMSC in patients unable or unwilling to undergo surgery. EBX and superficial radiation differ from conventional external-beam radiation. Patients typically undergo a course of 35 therapies in a seven-week period with a dose of 70Gg . Brachytherapy involves an external applicator positioned at a very short distance from its target. The accurate and precise placement of the applicator offers excellent results with minimized collateral damage. Brachtherapy may be new to dermatology, but it was already in use as a treatment for, among other things, prostate and breast cancer. Over time, high-dose radiation (HDR) was combined with EBX, which significantly reduced total radiation exposure (to 30 to 40Gg ) and resulted in fewer treatments for patients. This combination EBX with HDR was used to treat nonmelanoma skin cancers.
In a German study that commenced in 1987, 520 patients with skin cancer (BCC, SCC, Kaposi sarcoma, melanomas, skin manifestations of lymphomas, and solid organ tumors) were treated with 3,026 fraction using EBX with HDR.[187] Single doses of 5 to 10Gg were used once or twice a week with total doses of about 30 to 40Gg. In 91 percent of cases, complete remission was obtained, and in another six percent, partial remission was reported. Recurrence occurred in about eight percent of patients over 10 years. EBX with HDR was also used to treat 136 patients with facial BCC or SCC with a cure rate of 98 percent at five years and excellent cosmesis.[179] In fact, the local control rate for EBX with HDR is over 90 percent (Table 10).

A further evolution of ECB is the movement away from radioisotopes. HDR brachytherapy relies on iridium-192, while electronic brachytherapy (EBX) does not. By eliminating the radioisotope, EBX is potentially safer in that there is less collateral damage, even with the same high-dose fractionation; it is potentially more effective (beam is flatter with reduced penumbra), and it may be suitable for use within the clinic because it does not require a leaded vault. In terms of clinical workflow, a radiation oncologist, a radiation therapist, and a physicist are required. The patient enters the room and a portable shield is positioned; the machine is then calibrated and the dosing parameters are programmed. The patient is then treated. The entire visit takes about 15 minutes, and most protocols call for two treatments a week over the course of 8 to 10 weeks, but there is some variation in actual clinical practice. This method allows for the safe, effective use of EBR in a dermatology clinic, a physician’s office, or at a multidisciplinary care center. Data to date on EBX are promising. Bhatnagar presented one-year results from a study of 122 patients with 171 nonmelanoma skin cancers who were treated with HDR-EBX that demonstrated no recurrences at a mean follow up of 10 months (range 1–28 months).[197]
Later, Bhatnagar presented an update of this study where 187 patients with a total of 275 NMSC were treated with EBX.[197] The cancers were mostly BCC (n=159) and SCC (n=109). The surface applicator was placed 10, 20, 35, and 50mm distant with 2mm margin and depth of 3mm (the depth was determined by CT for particularly thick lesions). The dose fractionation was 5Gg x 8 fractions for a total of 40Gg , with treatments administered twice weekly. Adverse events were frequent (84% reported dermatitis, 25% pruritus) but were mostly mild. The most common late adverse event was hypopigmentation, which was mild and reported by 12 percent of participants. The vast majority of patients rated cosmesis with EBX as excellent (80% to 96%), and no recurrences were reported over 38 months.
The literature describes a multisite, multi-physician dermatology practice that introduced EBR for treating nonmelanoma skin cancer in the practice. After 15 months, the practice had treated 524 nonmelanoma skin cancer patients, and at 12.5 months of follow up, there were four recurrences. Cosmesis was excellent, and the practice was able to offer radiation therapy for certain patients as an alternative to MMS.[198] A group of physicians who used HDR-EBX in their practice to treat NMSC have reported treating 1,822 lesions from 2009 to 2014 with recurrence rates below one percent and excellent cosmesis. In this particular practice, lesions were mostly BCC or SCC (57% and 38%, respectively) and smaller than 2cm.[199]
Superficial radiation is a different treatment model based on low-energy photons that are able to penetrate up to 1cm. The equipment delivers a very focused beam, which reduces collateral tissue damage. Treatments last 90 seconds, and patients may need 5 to 22 fractions, depending on the size, site, and type of tumor involved. While a radiation therapist and a room with lead walls is required, superficial radiation can be offered in an office environment. In a retrospective study from 2000 to 2010 from a single office setting, 1,715 tumors in patients over 65 years of age and treated with superficial radiation were reviewed.200 Many of the patients had multiple tumors. Patients were offered the choice between MMS or superficial radiation; if they opted for superficial radiation, they received 5 to 7 fractions for a total of 35Gg . Patients were followed for a mean of 31.5 months (range 1–120 months) with an overall recurrence rate of 1.9 and five percent at 2 to 5 years. Recurrence rates were similar for BCC and SCC at two and 1.8 percent, respectively. The higher recurrence rates occurred in men and in tumors greater than 2cm in size.
In summary, HDR-EBX and superficial radiation offer excellent results although they are not quite equivalent to results obtainable with MMS. However, HDR-EBX and superficial radiation may be particularly appropriate for certain patients, namely those with larger tumors or tumors in very cosmetically sensitive areas (such as an eyelid), those with tumors in areas that typically heal slowly or poorly (such as the shin), and those taking blood thinners, are not suitable surgery candidates (older, frail patients), or who reject surgery as an option. It must be noted that the data presented are limited by the fact that there are not many large studies, and aggressive tumors are generally excluded from HDR-EBX and superficial radiation treatment. HDR has been studied since 1984, and some of the studies had long follow-up times, up to or surpassing five years. Dosing over the therapeutic range of EBX is comparable to isotope-based skin brachytherapy. Beyond the clinically useful range (>15mm), the 50kV EBX machine is lower, which serves to reduce the dose to nontarget structures. Superficial radiation has been around a long time, but there are fewer studies. A large prospective European study is being proposed that will compare EBX to MMS in multiple centers. A United States study is being conducted that combines EBX with confocal microscopy to enhance margin control.
Medicare reimburses EBX using a temporary code and simulation codes, but most private insurance payers do not reimburse EBX for skin cancer. Superficial radiation uses destruction codes. EBX reimbursement, even when available, has declined by about 75 percent in recent years. This has led to economic measures to make EBX more financially viable. Some companies are offering EBX equipment on a “pay-per-click” basis rather than requiring the practice to purchase the equipment outright; this allows clinics to pay only when the equipment is actually used. A superficial radiation machine may be set up to be run by a dermatologist and thus avoid the added costs of a radiation oncologist. Finally, practices that seek to add this specialized equipment are well advised to conduct budget analyses to be sure there are sufficient patients to support it.
With today’s patient-centric healthcare model, the reasons for selecting EBX over MMS are increasingly clear, particularly since the cure rates are similar. For this reason, it may be anticipated that EBX will gain in prominence in the coming years.
The ultimate goal in dermatology and medicine in general is personalized treatment, all the way to molecular targeting. The next step in the EBX evolution will be minimally invasive radiation therapy. To that end, much is needed: more prospective, long-term studies, more multidisciplinary conferences to share experiences, and the ability to incorporate treatment protocols for skin cancer into guidelines offered by national specialty societies. It would also be useful to define an “area under the curve” (AUC) for radiation therapy to better quantify treatments and compare data. Ideally, there should be a method where dermatologists can perform HDR-EBX and superficial radiation therapy independently, as dermatologists have traditionally been and continue to be skin cancer experts. Historically, too, dermatologists have typically performed radiation therapy on their patients with skin cancer. Thus, dermatologists need to be better represented in these therapies and participate in conferences and development of guidelines for HDR-EBX and superficial radiation. It may be that, in some areas, a hybrid system can emerge, bringing other disciplines to the care of skin cancer along with dermatologists.
The Management of SCC in the Immunocompromised Patient
Using a case study as an example, consider a 68-year-old white, female, immunosuppressed patient with multiple SCCs on her legs and at significant risk for current or future morbidity and mortality from skin cancer. The treatment goal is to prevent morbidity from her multiple primary tumors while simultaneously decreasing her mortality risk from high-risk tumors. Systemic retinoids may be appropriate for patients with multiple SCC when there is a high risk for metastatic cancer with SCC (risk less than or equal to 20%), when metastatic cancer is already present, when the patient is experiencing surgical fatigue, or when the patient’s pharmacological regimen calls for a decrease in immunosuppressive drugs. Systemic retinoids should be started at a low dose (10mg/day) and increased gradually up to 25mg/day, titrating the dose carefully to manage side effects. Systemic retinoids represent a possible life-long therapy for such patients, so it is important to manage adverse events. Occasional drug holidays may help patients cope with transient side effects. Adverse events associated with systemic retinoids may be grouped in three broad categories: The mucocutaneous side effects include skin peeling, cheilitis, and scalp alopecia; patients may experience musculoskeletal adverse events, including hyperostosis; and finally, systemic retinoids may cause or exacerbate hyperlipidemia, which may be managed with lipid-lowering agents. Patients should be advised that stopping treatment with systemic retinoids will increase their skin cancers.
Patients on systemic retinoids should be subjected to laboratory tests at baseline, monthly for the first three or four months of therapy, and then every three or four months thereafter once the patient is stable. These labs should include triglyceride monitoring and liver function testing.
As with any therapy, systemic retinoid treatment offers risks and benefits. The advantages include possible decrease of SCC and BCC and possible reduction of the risk for metastasis and recurrence. On the other hand, systemic retinoid is a life-long therapy that may not be well tolerated by patients. Systemic retinoid treatment is preventative rather than therapeutic and is associated with potentially serious side effects. In this particular case study, the 68-year-old patient could not afford the $800 a month it would cost for her to get acitretin. For such patients, ALA-PDT every 6 to 8 weeks may help prevent new lesions and may be reimbursable.
PDT can help reduce new SCC lesions in immunocompromised patients. In a study of solid organ transplant recipients treated with cyclic PDT, SCC lesions decreased 79 percent over baseline (73.3 to 81.8) in 12 months and 95 percent in 24 months (87.5% to 100%)201 The problem with PDT over the long term is that patients may grow weary of it and want to discontinue. Nevertheless, it represents a viable and important option.
Conclusion
This year’s MauiDerm convention continues to explore the new drugs, therapies, and approaches to psoriasis and skin cancers. The pathways associated with psoriasis continue to become better defined and with that, more effective and better tolerated treatments emerge.
References
- Strober B, Bagel J, Lebwohl M, et al. Efficacy and safety of apremilast in patients with moderate plaque psoriasis with lower BSA: week 16 results from the UNVEIL study. J Drugs Dermatol. 2017;16(8):801–808.
- Gottlieb AB, et al Certolizumab pegol treatment for chronic plaque psoriasis: 16-week primary results from two phase III, multicenter, randomized, placebo-controlled studies. Abstract 5077. Presented at American Academy of Dermatology 2017 Annual Meeting; 2017 Mar 4.
- Final Cimzia (certolizumab pegol) phase 3 trial meets primary efficacy endpoint in patients with moderate to severe chronic plaque psoriasis [news release]. Dermira; 2017 Jan 18. http://investor.dermira.com/phoenix.zhtml?c=253686&p=irol-newsArticle&ID=2238284
- Warren RB, Mrowietz U, von Kiedrowski R, et al. An intensified dosing schedule of subcutaneous methotrexate in patients with moderate to severe plaque-type psoriasis (METOP): a 52 week, multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10068):528–537.
- Wu JJ, Guerin A, Sundaram M, Dea K, Cloutier M, Mulani P. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76(1):81–90.
- Reich K, Papp KA2, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–288.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–417.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad of Dermatol. 2017;76(3):418–431.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol. 2015;136(1):116–124 e117.
- Lonnberg AS, Zachariae C, Skov L. Targeting of interleukin-17 in the treatment of psoriasis. Clin Cosmet Investig Dermatol. 2014;7:251–259.
- Jancin B. Secukinumab for psoriasis at 4 years: undiminished efficacy and safety. 18 Oct 2016; http://www.mdedge.com/edermatologynews/article/115936/psoriasis/secukinumab-psoriasis-4-years-undiminished-efficacy-and. Accessed April 30, 2017.
- Gordon KB, Blauvelt A, Papp KA, et al. Phase 3 Trials of Ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375(4):345–356.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373(14):1318–1328.
- Background Package for BLA 761032 Siliq (brodalumab) injection, 210 mg/1.5 ml [FDA briefing document]. Silver Spring, MD: Food and Drug Administration Center for Drug Evaluation and Research Office of New Drugs; 19 Jul 2016. https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/dermatologicandophthalmicdrugsadvisorycommittee/ucm511357.pdf. Accessed 30 Apr 2017.
- Gupta MA, Schork NJ, Gupta AK, Kirkby S, Ellis CN. Suicidal ideation in psoriasis. Int J Dermatol. 1993;32(3):188–190.
- Armstrong AW, Siegel MP, Bagel J, et al. From the medical board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76(2):290–298.
- Citrome L, Ketter TA. When does a difference make a difference? Interpretation of number needed to treat, number needed to harm, and likelihood to be helped or harmed. Int J Clin Pract. 2013;67(5):407–411.
- Kristensen LE, Jorgensen TS, Christensen R, et al. Societal costs and patients’ experience of health inequities before and after diagnosis of psoriatic arthritis: a Danish cohort study. Ann Rheum Dis. 2017;76(9):1495–1501.
- Spelman L, Su JC, Fernandez-Penas P, et al. Frequency of undiagnosed psoriatic arthritis among psoriasis patients in Australian dermatology practice. J Eur Acad Dermatol Venerol. 2015;29(11):2184–2191.
- Faustini F, Simon D, Oliveira I, et al. Subclinical joint inflammation in patients with psoriasis without concomitant psoriatic arthritis: a cross-sectional and longitudinal analysis. Ann Rheum Dis. 2016;75(12):2068–2074.
- Antonelli A, Ferrari SM, Giuggioli D, Ferrannini E, Ferri C, Fallahi P. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun Rev. 2014;13(3):272–280.
- Abji F, Pollock RA, Liang K, Chandran V, Gladman DD. Brief report: CXCL10 is a possible biomarker for the development of psoriatic arthritis among patients with psoriasis. Arthritis Rheumatol. 2016;68(12):2911-2916.
- Lewinson RT, Vallerand IA, Lowerison MW, et al. Depression is associated with an increased risk of psoriatic arthritis among patients with psoriasis: a population-based study. J Invest Dermatol. 2017;137(4):828–835.
- Tillett W, de-Vries C, McHugh NJ. Work disability in psoriatic arthritis: a systematic review. Rheumatology (Oxford). 2012;51(2):275–283.
- Tillett W, Shaddick G, Jobling A, et al. Effect of anti-TNF and conventional synthetic disease-modifying anti-rheumatic drug treatment on work disability and clinical outcome in a multicentre observational cohort study of psoriatic arthritis. Rheumatology (Oxford). 2017;56(4):603–612.
- 26. Iannone F, Lopalco G, Rigante D, Orlando I, Cantarini L, Lapadula G. Impact of obesity on the clinical outcome of rheumatologic patients in biotherapy. Autoimmun Rev. 2016;15(5):447–450.
- Di Lernia V, Tasin L, Pellicano R, Zumiani G, Albertini G. Impact of body mass index on retention rates of anti-TNF-alfa drugs in daily practice for psoriasis. J Dermatolog Treat. 2012;23(6):404–409.
- Hojgaard P, Glintborg B, Kristensen LE, Gudbjornsson B, Love TJ, Dreyer L. The influence of obesity on response to tumour necrosis factor-alpha inhibitors in psoriatic arthritis: results from the DANBIO and ICEBIO registries. Rheumatology (Oxford). 2016;55(12):2191–2199.
- Kirkham BW, Kavanaugh A, Reich K. Interleukin-17A: a unique pathway in immune-mediated diseases: psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology. 2014;141(2):133–142.
- Craven J. Sustained improvements in PsA domains with secukinumab. 14 Nov 2016; http://www.clinicaladvisor.com/acr-2016-coverage/three-year-clinical-improvement-in-psoriatic-arthritis-with-secukinumab/article/572636/. Accessed 2 May 2017.
- Mease P, Genovese MC, Gladstein G, et al. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. Arthritis Rheum. 2011;63(4):939–948.
- Ghoreschi K, Gadina M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp Dermatol. 2014;23(1):7–11.
- Mease P, Hall S, Fitzgerald O, et al. Efficacy and safety of tofacitinib, an oral janus kinase inhibitor, or adalimumab in patients with active psoriatic arthritis and an inadequate response to conventional synthetic dmards: a randomized, placebo-controlled, Phase 3 trial. Abstract 2983. Presented at the American College of Rheumatology Meeting; 15 Nov 2016.
- Gladman D, Rigby W, Azevedo V, et al. Efficacy and Safety of tofacitinib, an oral janus kinase inhibitor, in patients with active psoriatic arthritis and an inadequate response to tumor necrosis factor inhibitors: OPAL beyond, a randomized, double-blind, placebo-controlled, Phase 3 trial. Abstract 10L. American College of Rheumatology Meeting; 15 Nov 2016.
- Fowler JF, Jr., Hebert AA, Sugarman J. DFD-01, a novel medium potency betamethasone dipropionate 0.05% emollient spray, demonstrates similar efficacy to augmented betamethasone dipropionate 0.05% lotion for the treatment of moderate plaque psoriasis. J Drugs Dermatol. 2016;15(2):154–162.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis. J Am Acad Dermatol. 2009;60(4):643–659.
- Lebwohl M, Siskin SB, Epinette W, et al. A multicenter trial of calcipotriene ointment and halobetasol ointment compared with either agent alone for the treatment of psoriasis. J Am Acad of Dermatol. 1996;35(2 Pt 1):268–269.
- Lebwohl M, Yoles A, Lombardi K, Lou W. Calcipotriene ointment and halobetasol ointment in the long-term treatment of psoriasis: effects on the duration of improvement. J Am Acad of Dermatol. 1998;39(3):447–450.
- Kaufmann R, Bibby AJ, Bissonnette R, et al. A new calcipotriol/betamethasone dipropionate formulation (Daivobet) is an effective once-daily treatment for psoriasis vulgaris. Dermatology. 2002;205(4):389–393.
- Kubin ME, Kokkonen N, Palatsi R, et al. Clinical efficiency of topical calcipotriol/betamethasone treatment in psoriasis relies on suppression of the inflammatory TNFalpha – IL-23 – IL-17 axis. Acta Derm Venereol. 2017;97(4):449–455.
- Koo J, Tyring S, Werschler WP, et al. Superior efficacy of calcipotriene and betamethasone dipropionate aerosol foam versus ointment in patients with psoriasis vulgaris – A randomized phase II study. J Dermtolog Treat. 2016;27(2):120–127.
- Lebwohl MG, Breneman DL, Goffe BS, et al. Tazarotene 0.1% gel plus corticosteroid cream in the treatment of plaque psoriasis. J Am Acad Dermatol. 1998;39(4 Pt 1):590–596.
- Sugarman JL, Gold LS, Lebwohl MG, Pariser DM, Alexander BJ, Pillai R. A Phase 2, multicenter, double-blind, randomized, vehicle controlled clinical study to assess the safety and efficacy of a halobetasol/tazarotene fixed combination in the treatment of plaque psoriasis. J Am Acad Dermatol. 2017;16(3):197–204.
- Mavers M, Ruderman EM, Perlman H. Intracellular signal pathways: potential for therapies. Curr Rheumatol Rep. 2009;11(5):378–385.
- Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7(3):191–201.
- Ghoreschi K, Jesson MI, Li X, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). Journal Immunol. 2011;186(7):4234–4243.
- Shuai K, Liu B. Regulation of JAK-STAT signaling in the immune system. Nat Rev Immunol. 2003;3(11):900–911.
- O’Sullivan LA, Liongue C, Lewis RS, Stephenson SE, Ward AC. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol Immunol. 2007;44(10):2497–2506.
- O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36(4):542–550.
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–442.
- Papp KA, Bissonnette R, Gooderham M, et al. Treatment of plaque psoriasis with an ointment formulation of the Janus kinase inhibitor, tofacitinib: a Phase 2b randomized clinical trial. BMC Dermatol. 2016;16(1):15.
- Punwani N, Burn T, Scherle P, et al. Downmodulation of key inflammatory cell markers with a topical Janus kinase 1/2 inhibitor. Br J Dermatol. 2015;173(4):989–997.
- Christophers E. Comorbidities in psoriasis. Clin Dermatol. 2007;25(6):529–534.
- Egeberg A, Skov L, Joshi AA, et al. The relationship between duration of psoriasis, vascular inflammation, and cardiovascular events. J Am Acad Dermatol. 2017;S0190–9622(17)31925–4.
- Tula E, Ergun T, Seckin D, Ozgen Z, Avsar E. Psoriasis and the liver: problems, causes and course. The Australas J Dermatol. 2017; 58(3):194–199.
- Jensen P, Egeberg A, Gislason G, Hansen PR, Thyssen JP, Skov L. Increased risk of autoimmune hepatitis in patients with psoriasis: a Danish nationwide cohort study. J Invest Dermatol. 2016;136(7):1515–1517.
- Braganza J, Lee S, McCloy R, McMahon M. Chronic pancreatitis. Lancet. 2011;377:1184–1197.
- Chiu HY, Hsieh CF, Chiang YT, Huang WF, Tsai TF. The risk of chronic pancreatitis in patients with psoriasis: a population-based cohort study. PLoS One. 2016;11(7):e0160041.
- Chiu HY, Wang IT, Huang WF, Tsai YW, Shiu MN, Tsai TF. Increased risk of avascular necrosis in patients with psoriatic disease: a nationwide population-based matched cohort study. J Am Acad Dermatol. 2017;76(5):903–910.e901.
- Gelfand JM, Troxel AB, Lewis JD, et al. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol. 2007;143(12):1493–1499.
- Prodanovich S, Kirsner RS, Kravetz JD, Ma F, Martinez L, Federman DG. Association of psoriasis with coronary artery, cerebrovascular, and peripheral vascular diseases and mortality. Arch Dermatol. 2009;145(6):700–703.
- Balci DD, Balci A, Karazincir S, et al. Increased carotid artery intima-media thickness and impaired endothelial function in psoriasis. J Eur Acad of Dermatol and Venereol. 2009;23(1):1–6.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6-month prospective study. J Dermatol. 2016;43(11):1267–1272.
- Hjuler KF, Bottcher M, Vestergaard C, Botker HE, Iversen L, Kragballe K. Association between changes in coronary artery disease progression and treatment with biologic agents for severe psoriasis. JAMA Dermatol. 2016;152(10):1114–1121.
- Wu JJ, Guerin A, Sundaram M, Dea K, Cloutier M, Mulani P. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-alpha inhibitors versus methotrexate. J Am Acad Dermatol. 2017;76(1):81–90.
- Bos JD, Hulsebosch HJ, Krieg SR, Bakker PM, Cormane RH. Immunocompetent cells in psoriasis. In situ immunophenotyping by monoclonal antibodies. Arch Dermatol Res. 1983;275(3):181–189.
- Bos JD, Hagenaars C, Das PK, Krieg SR, Voorn WJ, Kapsenberg ML. Predominance of “memory” T cells (CD4+, CDw29+) over “naive” T cells (CD4+, CD45R+) in both normal and diseased human skin. Arch Dermatol Res. 1989;281(1):24–30.
- Gottlieb SL, Gilleaudeau P, Johnson R, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. 1995;1(5):442–447.
- Kaffenberger BH, Kaffenberger TM, Wong HK. Immunotargeting in the management of psoriasis. ImmunoTargets Ther. 2013;2:51–60.
- Ganguly D, Chamilos G, Lande R, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206(9):1983–1994.
- Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564–569.
- Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294(5546):1540–1543.
- Zaba LC, Krueger JG, Lowes MA. Resident and “inflammatory” dendritic cells in human skin. J Invest Dermatol. 2009;129(2):302–308.
- Lowes MA, Chamian F, Abello MV, et al. Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci U S A. 2005;102(52):19057–19062.
- Chamian F, Lowes MA, Lin SL, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A. 2005;102(6):2075–2080.
- Tonel G, Conrad C, Laggner U, et al. Cutting edge: a critical functional role for IL-23 in psoriasis. J Immunol. 2010;185(10):5688–5691.
- Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–234.
- Morishima N, Mizoguchi I, Takeda K, Mizuguchi J, Yoshimoto T. TGF-beta is necessary for induction of IL-23R and Th17 differentiation by IL-6 and IL-23. Biochem Biophys Res Commun. 2009;386(1):105–110.
- Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159(5):1092–1102.
- Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. 2011;131(3):677–687.
- Krueger JG, Fretzin S, Suarez-Farinas M, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012;130(1):145–154 e149.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150(3):407–415.
- Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174(6):3695–3702.
- Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8(9):950–957.
- Zhang W, Dang E, Shi X, et al. The pro-inflammatory cytokine IL-22 up-regulates keratin 17 expression in keratinocytes via STAT3 and ERK1/2. PloS One. 2012;7(7):e40797.
- Oestreicher JL, Walters IB, Kikuchi T, et al. Molecular classification of psoriasis disease-associated genes through pharmacogenomic expression profiling. Pharmacogenomics J. 2001;1(4):272–287.
- Suarez-Farinas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG. Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J Invest Dermatol. 2012;132(11):2552–2564.
- Wolk K, Witte E, Wallace E, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36(5):1309–1323.
- Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21(2):241–254.
- Sa SM, Valdez PA, Wu J, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. 2007;178(4):2229–2240.
- Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. The British journal of dermatology. 2008;159(5):1092–1102.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. New Engl J Med. 2009;361(5):496–509.
- Kauffman C, Aria N, Toichi E, et al. A phase 1 study evaluating the safety, pharmacokinetics, and clinical response of a human IL-12 p40 antibody in subjects with plaque psoriasis. J Invest Dermatol. 2004;123:1037–1044.
- Leonardi CL, Kimball AB, Papp KA, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. 2008;371(9625):1665–1674.
- Papp KA, Langley RG, Lebwohl M, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. 2008;371(9625):1675–1684.
- Harden J, Johnson-Huang L, Chamian M, et al. Humanized anti-IFN-gamma (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol. 2015;135(2):553–6.
- Amatya N, Garg AV, Gaffen SL. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017;38(5):310–322.
- Gaffen SL. Structure and signaling in the IL-17 receptor family. Nat Rev Immunol. 2009;9(8):556–567.
- Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2(52):52ra72.
- Papp KA, Reid C, Foley P, et al. Anti-IL-17 receptor antibody AMG 827 leads to rapid clinical response in subjects with moderate to severe psoriasis: results from a phase I, randomized, placebo-controlled trial. J Invest Dermatol. 2012;132(10):2466–2469.
- Russell CB, Rand H, Bigler J, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. 2014;192(8):3828–3836.
- Leonardi C, Matheson R, Zachariae C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366(13):1190–1199.
- Puig L. The role of IL 23 in the treatment of psoriasis. Expert Rev Clin Immunol. 2017;13(6):525–534.
- Nawas Z, Hatch M, Ramos E, et al. A review of guselkumab, an IL-23 inhibitor, for moderate-to-severe plaque psoriasis. Skin Therapy Lett. 2017;22(2):8–10.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76(3):418–431.
- Reichert JM. Antibodies to watch in 2017. MAbs. 2017;9(2):167–181.
- Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376(16):1551–1560.
- Kim J, Krueger JG. Highly effective new treatments for psoriasis target the IL-23/Type 17 T cell autoimmune axis. Annu Rev Med. 2017;68:255–269.
- Kim J, Oh CH, Jeon J, et al. Molecular phenotyping small (Asian) versus large (Western) plaque psoriasis shows common activation of IL-17 pathway genes but different regulatory gene sets. J Invest Dermatol. 2016;136(1):161–172.
- Gelfand JM, Stern RS, Nijsten T, et al. The prevalence of psoriasis in African Americans: results from a population-based study. J Am Acad Dermatol. 2005;52(1):23-26.
- Lande R, Botti E, Jandus C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. 2014;5:5621.
- Ganguly D, Chamilos G, Lande R, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206(9):1983–1994.
- Lande R, Gregorio J, Facchinetti V, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449(7162):564–569.
- Chamilos G, Gregorio J, Meller S, et al. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood. 2012;120(18):3699–3707.
- Nair RP, Stuart PE, Nistor I, et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. 2006;78(5):827–851.
- Arakawa A, Siewert K, Stohr J, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. 2015;212(13):2203–2212.
- Clark RA. Skin resident T cells: the ups and downs of on site immunity. J Invest Dermatol. 2010 Feb; 130(2): 362–370.
- Gulati N, Suarez-Farinas M, Correa da Rosa J, Krueger JG. Psoriasis is characterized by deficient negative immune regulation compared to transient delayed-type hypersensitivity reactions. F1000Res. 2015;4:149.
- American Cancer Society. Skin Cancer. 2017; https://www.cancer.org/cancer/skin-cancer.html. Accessed 21 Apr 2017.
- Nindl I, Gottschling M, Stockfleth E. Human papillomaviruses and non-melanoma skin cancer: basic virology and clinical manifestations. Dis Markers. 2007;23(4):247–259.
- Zentrum fur Krebsregisterdaten. Epidemiologische Krebsregistrierung in Deutschland. Krebs in Deutschland. 2008;.http://www.krebsdaten.de/Krebs/DE/Content/Publikationen/Krebs_in_Deutschland/kid_2015/kid_2015_epi_registrierung.pdf;jsessionid=6414E9AEEE7E73714E806E983D4FDE5B.1_cid381?__blob=publicationFile. Accessed 21 Apr 2017.
- Doorbar J. The papillomavirus life cycle. J Clin Virol. 2005;32:7–15
- Stockfleth E. The importance of treating the field in actinic keratosis. J Eur Acad Dermatol Venereol. 2017;31 Suppl 2:8–11
- Padilla RS, Sebastian S, Jiang Z, Nindl I, Larson R. Gene expression patterns of normal human skin, actinic keratosis, and squamous cell carcinoma: a spectrum of disease progression. Arch Dermatol. 2010;146(3):288–293.
- Fernandez-Figueras MT, Carrato C, Saenz X, et al. Actinic keratosis with atypical basal cells (AK I) is the most common lesion associated with invasive squamous cell carcinoma of the skin. J Eur Acad Dermatol Venereol. 2015;29(5):991–997.
- Stockfleth E, Hinrichs B, Surber C, Christophers E. Prevention initiative for dermatological malignancies: where do we stand? Br J Dermatol. 2012;167 Suppl 2:v–vi.
- Stockfleth E. Updates on the biology and treatment of actinic keratosis. Presented at the 13th Congress of the EADO; 2017 May 3–6.
- Meyer T, Surber C, French LE, Stockfleth E. Resiquimod, a topical drug for viral skin lesions and skin cancer. Expert Opin Investig Drugs. 2013;22(1):149–159.
- Hengge UR, Ruzicka T. Topical immunomodulation in dermatology: potential of toll-like receptor agonists. Dermatol Surg. 2004;30(8):1101–1112.
- Szeimies RM, Bichel J, Ortonne JP, Stockfleth E, Lee J, Meng TC. A phase II dose-ranging study of topical resiquimod to treat actinic keratosis. Br J Dermatol. 2008;159(1):205–210.
- Lebwohl M, Shumack S, Stein Gold L, Melgaard A, Larsson T, Tyring SK. Long-term follow-up study of ingenol mebutate gel for the treatment of actinic keratoses. JAMA Dermatol. 2013;149(6):666–670.
- Lebwohl M, Swanson N, Anderson LL, Melgaard A, Xu Z, Berman B. Ingenol mebutate gel for actinic keratosis. N Engl J Med. 2012;366(11):1010–1019.
- Ogbourne SM, Suhrbier A, Jones B, et al. Antitumor activity of 3-ingenyl angelate: plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004;64(8):2833–2839.
- Challacombe JM, Suhrbier A, Parsons PG, et al. Neutrophils are a key component of the antitumor efficacy of topical chemotherapy with ingenol-3-angelate. J Immunol. 2006;177(11):8123–8132.
- Ersvaer E, Kittang AO, Hampson P, et al. The protein kinase C agonist PEP005 (ingenol 3-angelate) in the treatment of human cancer: a balance between efficacy and toxicity. Toxins (Basel). 2010;2(1):174–194.
- Berman B, Amini S, Valins W, Block S. Pharmacotherapy of actinic keratosis. Expert Opin Pharmacother. 2009;10(18):3015–3031.
- Hampson P, Kavanagh D, Smith E, Wang K, Lord JM, Ed Rainger G. The anti-tumor agent, ingenol-3-angelate (PEP005), promotes the recruitment of cytotoxic neutrophils by activation of vascular endothelial cells in a PKC-delta dependent manner. Cancer Immunol Immunother. 2008;57(8):1241–1251.
- Schlaak M, Simon JC. Topical treatment of actinic keratoses with low-dose 5-fluorouracil in combination with salicylic acid–pilot study. J Dtsch Dermatol Ges. 2010;8(3):174–178.
- Stockfleth E, Kerl H, Zwingers T, Willers C. Low-dose 5-fluorouracil in combination with salicylic acid as a new lesion-directed option to treat topically actinic keratoses: histological and clinical study results. Br J Dermatol. 2011;165(5):1101–1108.
- Stockfleth E, Zwingers T, Willers C. Recurrence rates and patient assessed outcomes of 0.5% 5-fluorouracil in combination with salicylic acid treating actinic keratoses. Eur J Dermatol. 2012;22(3):370–374.
- Stockfleth E, von Kiedrowski R, Dominicus R, et al. Efficacy and safety of 5-fluorouracil 0.5%/salicylic acid 10% in the field-directed treatment of actinic keratosis: a Phase III, randomized, double-blind, vehicle-controlled trial. Dermatol Ther (Heidelb). 2017;7(1):81–96.
- Hanahan D. Rethinking the war on cancer. Lancet. 2014;383(9916):558–563.
- Schmitt JV, Chinem VP, Marques ME, Miot HA. Increase in the incidence of basal cell carcinoma in a university hospital between 1999 and 2009. An Bras Dermatol. 2011;86(2):375–377.
- van Hattem S, Aarts MJ, Louwman WJ, et al. Increase in basal cell carcinoma incidence steepest in individuals with high socioeconomic status: results of a cancer registry study in The Netherlands. Br J Dermatol. 2009;161(4):840–845.
- Asgari MM, Moffet HH, Ray GT, Quesenberry CP. Trends in basal cell carcinoma incidence and identification of high-risk subgroups, 1998-2012. JAMA Dermatol. 2015;151(9):976–981.
- American Cancer Society. Key statistics for basal and squamous cell skin cancers. 10 May 2016; https://www.cancer.org/cancer/basal-and-squamous-cell-skin-cancer/about/key-statistics.html. Accessed 7 May 2017.
- Walko CM, Lindley C. Capecitabine: a review. Clin Ther. 2005;27(1):23–44.
- Weiss JM, Bagley S, Hwang WT, et al. Capecitabine and lapatinib for the first-line treatment of metastatic/recurrent head and neck squamous cell carcinoma. Cancer. 2016;122(15):2350–2355.
- Marmur ES, Schmults CD, Goldberg DJ. A review of laser and photodynamic therapy for the treatment of nonmelanoma skin cancer. Dermatol Surg. 2004;30(2 Pt 2):264–271.
- Dragieva G, Hafner J, Dummer R, et al. Topical photodynamic therapy in the treatment of actinic keratoses and Bowen’s disease in transplant recipients. Transplantation. 2004;77(1):115–121.
- Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore). 1987;66(2):98–113.
- Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet. 2001;10(7):757-762.
- Oseroff AR, Shieh S, Frawley NP, et al. Treatment of diffuse basal cell carcinomas and basaloid follicular hamartomas in nevoid basal cell carcinoma syndrome by wide-area 5-aminolevulinic acid photodynamic therapy. Arch Dermatol. 2005;141(1):60–67.
- Zeitouni NC, Sunar U, Rohrbach DJ, et al. A prospective study of pain control by a 2-step irradiance schedule during topical photodynamic therapy of nonmelanoma skin cancer. Dermatol Surg. 2014;40(12):1390–1394.
- Schmitz L, Novak B, Hoeh AK, Luebbert H, Dirschka T. Epidermal penetration and protoporphyrin IX formation of two different 5-aminolevulinic acid formulations in ex vivo human skin. Photodiagnosis Photodyn Ther. 2016;14:40–46.
- Ameluz [assessment report]. European Medicines Agency Committee for Medicinal Products for Human Use; 15 Dec 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/002204/WC500222131.pdf
- Hengge UR, Schaller J. Successful treatment of invasive squamous cell carcinoma using topical imiquimod. Arch Dermatol. 2004;140(4):404–406.
- Kossard S. Treatment of large facial Bowen’s disease: case report. Clin Exp Dermatol. 2003;28 Suppl 1:13–15.
- Ondo AL, Mings SM, Pestak RM, Shanler SD. Topical combination therapy for cutaneous squamous cell carcinoma in situ with 5-fluorouracil cream and imiquimod cream in patients who have failed topical monotherapy. J Am Acad Dermatol. 2006;55(6):1092–1094.
- McKay KM, Sambrano BL, Fox PS, Bassett RL, Chon S, Prieto VG. Thickness of superficial basal cell carcinoma (sBCC) predicts imiquimod efficacy: a proposal for a thickness-based definition of sBCC. Br J Dermatol. 2013;169(3):549–554.
- Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol. 2009;145(12):1431–1438.
- Bettencourt MS. Treatment of superficial basal cell carcinoma with ingenol mebutate gel, 0.05%. Clin Cosmet Investig Dermatol. 2016;9:205–209.
- Kirby JS, Miller CJ. Intralesional chemotherapy for nonmelanoma skin cancer: a practical review. J Am Acad Dermatol. 2010;63(4):689–702.
- Arpey C, Annest N, Tucker S, Rapini R, MacFarlane D. Intralesional and Perilesional Treatment of Skin Cancers. Skin Cancer Management: Springer Science + Business Media, LLC; 2010:57–76.
- Longo C, Ragazzi M, Rajadhyaksha M, et al. In Vivo and Ex Vivo Confocal Microscopy for Dermatologic and Mohs Surgeons. Dermatol Clin. 2016;34(4):497–504.
- Shin TM, Etzkorn JR, Sobanko JF, et al. Clinical factors associated with subclinical spread of in situ melanoma. J Am Acad Dermatol. 2017;76(4):707–713.
- Shin TM, Shaikh WR, Etzkorn JR, et al. Clinical and pathologic factors associated with subclinical spread of invasive melanoma. J AM Acad Dermatol. 2017;76(4):714–721.
- Moyer JS, Rudy S, Boonstra PS, et al. Efficacy of staged excision with permanent section margin control for cutaneous head and neck melanoma. JAMA Dermatol (Chicago, Ill). 2017;153(3):282–288.
- Friedman BJ, Pimentel JD, Ozog DM. Achieving optimal en face tissue sections during staged lentigo maligna excisions: a novel technique. Dermatol Surg. 2015;41(11):1332–1335.
- Bricca G, Brodland D, Zitelli J. Immunostaining melanoma frozen sections: the 1-hour protocol. Dermatol Surg. 2004;30:403–408.
- Christensen KN, Hochwalt PC, Hocker TL, et al. Comparison of MITF and melan-a immunohistochemistry during mohs surgery for lentigo maligna-type melanoma in situ and lentigo maligna melanoma. Dermatol Surg. 2016;42(2):167–175.
- Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 Blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. 2016;374(26):2542–2552.
- Iyer JG, Blom A, Doumani R, et al. Response rates and durability of chemotherapy among 62 patients with metastatic Merkel cell carcinoma. Cancer Med. 2016;5(9):2294–2301.
- Lebbe C, Becker J, Grob J. Diagnosis and treatment of Merkel cell carcinoma. European consensus-based interdisciplinary guideline. Eur J Cancer. 2015;51:2396–2403.
- Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–1385.
- Winkler JK, Dimitrakopoulou-Strauss A, Sachpekidis C, Enk A, Hassel JC. Ipilimumab has efficacy in metastatic Merkel cell carcinoma: a case series of five patients. J Eur Acad Dermatol Venereol. 2017.
- Pariser DM, Houlihan A, Ferdon MB, Berg JE. Randomized vehicle-controlled study of short drug incubation aminolevulinic acid photodynamic therapy for actinic keratoses of the face or scalp. Dermatol Surg. 2016;42(3):296–304.
- Nissen CV1, Heerfordt IM, Wiegell SR, Mikkelsen CS, Wulf HC. Pretreatment with 5-fluorouracil cream enhances the efficacy of daylight-mediated photodynamic therapy for actinic keratosis. Acta Derm Venereol. 2017;97(5):617–621.
- Willey A, Anderson RR, Sakamoto FH. Temperature-modulated photodynamic therapy for the treatment of actinic keratosis on the extremities: a one-year follow-up study. Dermatol Surg. 2015;41(11):1290–1295.
- Ozog DM, Rkein AM, Fabi SG, et al. Photodynamic therapy: a clinical consensus guide. Dermatol Surg. 2016;42(7):804–827.
- Martin GM. In-office painless aminolevulinic acid photodynamic therapy: a proof of concept study and clinical Experience in more than 100 patients. J Clin Aesthet Dermatol. 2016;9(2):19–26.
- Reinhold U, Dirschka T, Ostendorf R, et al. A randomized, double-blind, phase III, multicentre study to evaluate the safety and efficacy of BF-200 ALA (Ameluz((R)) ) vs. placebo in the field-directed treatment of mild-to-moderate actinic keratosis with photodynamic therapy (PDT) when using the BF-RhodoLED((R)) lamp. Br J Dermatol. 2016;175(4):696–705.
- Dirschka T, Radny P, Dominicus R, et al. Photodynamic therapy with BF-200 ALA for the treatment of actinic keratosis: results of a multicentre, randomized, observer-blind phase III study in comparison with a registered methyl-5-aminolaevulinate cream and placebo. Br J Dermatol. 2012;166(1):137–146.
- Piacquadio D, Houlihan A, Ferdon M, Berg JE, Marcus SM. Blue light photodynamic therapy (PDT) with aminolevulinic acid (ALA) 20% reduces occurrence of acitinic keratoses (AK) and de novo non-melanoma skin cancer (NMSC) in high risk patients. Presented at MauiDerm 2017; 2017 Mar 20; Maui, Hawaii.
- Jorizzo J, Stewart D, Bucko A, et al. Randomized trial evaluating a new 0.5% fluorouracil formulation demonstrates efficacy after 1-, 2-, or 4-week treatment in patients with actinic keratosis. Cutis. 2002;70(6):335–339.
- Weiss J, Menter A, Hevia O, et al. Effective treatment of actinic keratosis with 0.5% fluorouracil cream for 1, 2, or 4 weeks. Cutis. 2002;70(2 Suppl):22–29.
- Kohler-Brock A, Prager W, Pohlmann S, Kunze S. [The indications for and results of HDR afterloading therapy in diseases of the skin and mucosa with standardized surface applicators (the Leipzig applicator)]. Strahlenther Onkol. 1999;175(4):170–174.
- Guix B, Finestres F, Tello J, et al. Treatment of skin carcinomas of the face by high-dose-rate brachytherapy and custom-made surface molds. Int J Radiat Oncol Biol Phys. 2000;47(1):95–102.
- Maes A, Garmyn M, Stas M, Van Limbergen E. [The treatment of basal and squamous cell carcinomas of the skin with brachytherapy]. Tijdschrift voor Geneeskunde. 2004;60(10):683–691.
- Debois JM. Cesium-137 brachytherapy for epithelioma of the skin of the nose: experience with 370 patients. J Belge Radiol. 1994;77(1):1–4.
- Crook JM, Mazeron JJ, Marinello G, et al. Interstitial iridium 192 for cutaneous carcinoma of the external nose. Int J Radiat Oncol Biol Phys. 1990;18(1):243–248.
- Mazeron JJ, Chassagne D, Crook J, et al. Radiation therapy of carcinomas of the skin of nose and nasal vestibule: a report of 1676 cases by the Groupe Europeen de Curiethérapie. Radiother Oncol. 1988;13(3):165–73.
- Daly NJ, de Lafontan B, Combes PF. Results of the treatment of 165 lid carcinomas by iridium wire implant. Results of the treatment of 165 lid carcinomas by iridium wire implant. Int J Radiat Oncol Biol Phys. 1984;10(4):455–459.
- Krengli M, Masini L, Comoli AM, et al. Interstitial brachytherapy for eyelid carcinoma. Outcome analysis in 60 patients. Strahlenther Onkol. 2014;190(3):245–9.
- Mazeron JJ, Ghalie R, Zeller J, et al. Radiation therapy for carcinoma of the pinna using iridium 192 wires: a series of 70 patients. Int J Radiat Oncol Biol Phys. 1986;12(10):1757–1763.
- Baris G, Visser AG, van Andel JG. The treatment of squamous cell carcinoma of the nasal vestibule with interstitial iridium implantation. Radiother Oncol. 1985;4(2):121–125.
- Bhatnagar A. Electronic brachytherapy for the treatment of nonmelanoma skin cancer: results at 3 years. Abstract 157. Presented at the American Society of Radiation Oncology’s (ASTRO) 55th Annual Meeting; 2013 September 22–25.
- Doggett S, Willoughby M, Willoughby C, Mafong E, Han A. Incorporation of electronic brachytherapy for skin cancer into a community dermatology practice. J Clin Aesthet Dermatol. 2015;8(11):28–32.
- Bhatnagar A, Patel R, Werschler WP, Ceilley RI, Strimling R. High-dose rate electronic brachytherapy: a nonsurgical treatment alternative for nonmelanoma skin cancer. J Clin Aesthet Dermatol. 2016;9(11):16–22.
- Cognetta AB, Howard BM, Heaton HP, Stoddard ER, Hong HG, Green WH. Superficial x-ray in the treatment of basal