J Clin Aesthet Dermatol. 2025;18(10):40–47.
by Beth Childs, MS, and Joseph F. Merola, MD, MMSc
Dr. Merola and Ms. Childs are with the Department of Dermatology, Department of Medicine, and Division of Rheumatology at UT Southwestern Medical Center in Dallas, Texas.
FUNDING: We would like to extend our sincere thanks to International Dermatology Outcome Measures (IDEOM) for supporting the Masterclass in Dermatology series.
DISCLOSURES: Dr. Merola serves on the medical and scientific board of the Lupus Foundation of America and is a consultant and/or investigator for AbbVie, Amgen, Astra Zeneca, Boehringer Inhelheim, Bristol-Myers Squibb, Dermavant, Eli Lilly, Incyte, Novartis, Janssen, UCB, Sanofi-Regeneron, Sun Pharma, Biogen, Pfizer and Leo Pharma. Ms. Childs has no conflicts to declare.
Abstract: Cutaneous lupus erythematosus (CLE) may occur independently or in association with systemic lupus erythematosus (SLE). When systemic disease is present, CLE is the first manifestation in nearly one-third of cases. This positions dermatologists as key stakeholders in early detection of systemic disease, underscoring the importance of appropriate screening among this population. Various CLE subtypes carry distinct risks of systemic progression, with acute CLE closely tied to active SLE, subacute CLE conferring moderate risk, and most chronic subtypes (eg, localized discoid lupus) remaining limited to the skin. This review provides a practical, dermatology-focused framework for risk stratification, screening, and comanagement of patients with CLE. To support clinical decision-making and expand awareness, we introduce the “LABS FOR” SLE mnemonic to guide laboratory evaluation and propose an updated visual algorithm that illustrates screening and monitoring practices. We synthesize evidence-based and expert-informed recommendations, including serologic, demographic, clinical, and genetic predictors of systemic involvement. The 2019 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria—requiring antinuclear antibody (ANA) positivity and weighted domain scoring—are reviewed and compared to other diagnostic aids. Additionally, we highlight appropriate ANA testing, the importance of symptom review, and targeted second-line labs. Finally, we discuss collaborative management strategies with rheumatology, including organ-specific therapeutic considerations. By adopting a structured, CLE-informed approach to systemic screening and follow-up, dermatologists can play a critical role in improving outcomes for patients across the lupus spectrum. Keywords. Cutaneous lupus erythematosus, systemic lupus erythematosus, screening, rheumatology, quality improvement, multidisciplinary care, risk stratification, mnemonic
Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease in which cutaneous involvement is both common and clinically significant. Cutaneous lupus erythematosus (CLE) occurs in up to 85% of patients with SLE and is the initial manifestation in approximately 29%, making dermatologists key players in early detection, risk stratification, and comanagement.1, 2 Recognizing CLE is essential, as skin lesions can be the first sign of systemic disease, significantly impact quality of life, cause disfiguring scarring, and, if not treated promptly, may precede irreversible organ damage.3
At the 2025 Masterclass in Dermatology in Sarasota, Florida, Dr. Joseph F. Merola emphasized the critical role of dermatologists in screening for SLE in patients with CLE, much like they screen for psoriatic arthritis in psoriasis. Dermatologists must not only recognize when rheumatology referral or co-management is appropriate but also feel confident managing the skin manifestations of CLE within the broader context of potential systemic disease. This review synthesizes key insights from that meeting and the latest literature to provide a comprehensive, practical framework for dermatologists. We first examine the transition from cutaneous to systemic lupus, including how CLE phenotypic subtypes are variably correlated with systemic disease risk, then outline predictors of progression to SLE, including serologic markers, genetics, and clinical features. We discuss the relevance of updated American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria, introduce a streamlined screening tool for busy clinical settings, and provide guidance for longitudinal monitoring. Finally, we present collaborative management strategies, including examples of how systemic therapies may be selected based on organ involvement in patients with both cutaneous and systemic disease.
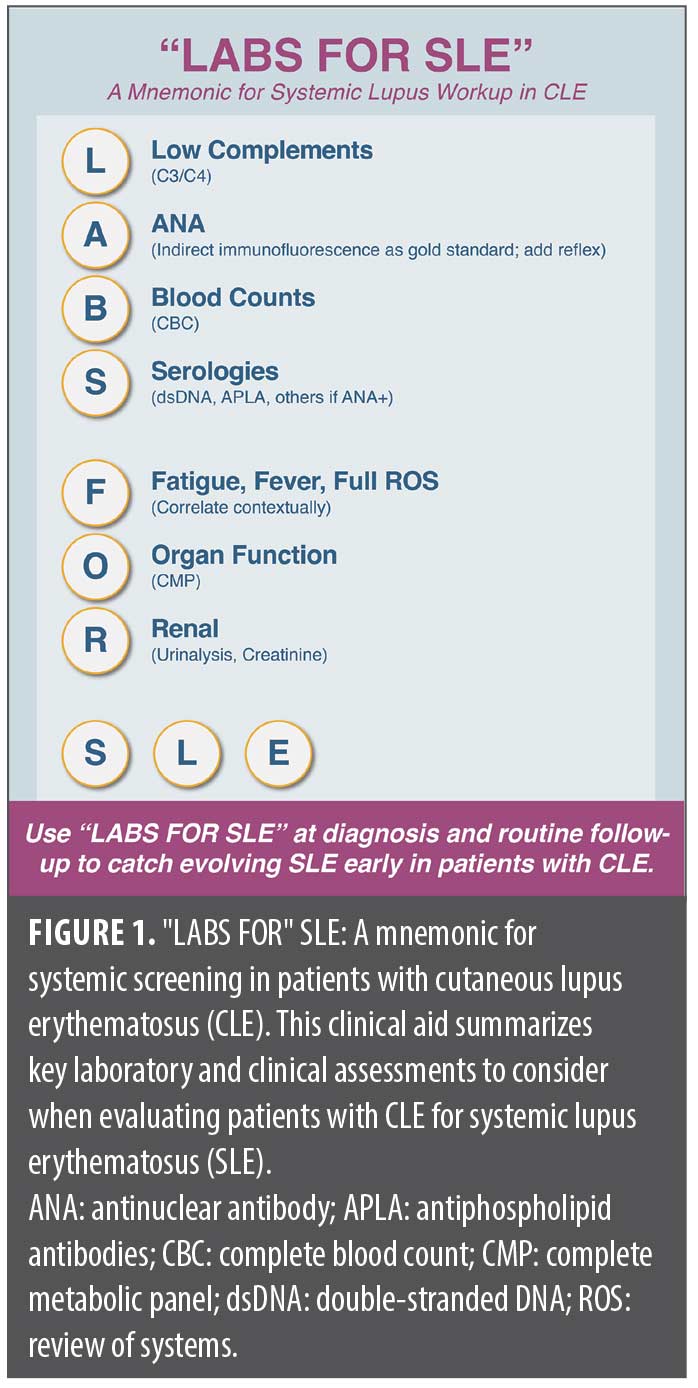
To promote systematic SLE assessment in patients with CLE and support durable awareness among trainees, we introduce a practical mnemonic—”LABS FOR” SLE (Low complements, Antinuclear antibody [ANA], Blood counts, Serologies, Fatigue/Fever/Full review of systems [ROS], Organ function, and Renal check)—as a streamlined tool to help dermatologists efficiently screen for systemic involvement. Next, we outline a more detailed, optimized approach to screening and monitoring patients with CLE, involving baseline laboratory workup, “red flag” symptoms, and surveillance strategies. Finally, we highlight best practices in dermatology-rheumatology collaboration, reinforcing the importance of multidisciplinary care. This article aims to provide a clear, practical, and up-to-date resource for dermatologists navigating the complexities of CLE and SLE.
Why Dermatologists Need to Be Well-Versed in SLE
Dermatologists are often the first specialists to diagnose SLE, as cutaneous manifestations frequently precede or accompany systemic disease. CLE is not rare; its incidence (3 to 4 per 100,000) and prevalence (approximately 70 per 100,000) are comparable to those of SLE.1 Up to 29% of patients with SLE first present with skin findings, underscoring the critical role of dermatologists in early detection and risk stratification.2
Beyond its diagnostic significance, CLE can be debilitating, causing pain, pruritus, scarring, and significant psychosocial distress.1,3 Prompt recognition and treatment are essential to control disease activity and prevent permanent scarring, particularly in chronic forms like discoid lupus. Importantly, CLE is not always limited to the skin; 10 to 25% of patients initially diagnosed with CLE progress to SLE, necessitating ongoing systemic evaluation.1,4 Early detection of systemic symptoms such as fever, arthritis, nephritis, or serositis can facilitate timely rheumatologic referral and prevent irreversible organ damage.
The diagnostic weight of skin findings in SLE is well established; four of the classic 1997 ACR criteria (malar rash, discoid rash, photosensitivity, oral ulcers) were cutaneous. While the 2019 ACR/EULAR criteria prioritize serologic markers, mucocutaneous features remain key to classification and management.5,6 Dermatologists must therefore be adept at diagnosing CLE, distinguishing subtypes, and recognizing systemic risk factors to ensure patients receive early intervention and multidisciplinary care.
A Mnemonic for Systematic SLE Assessment in CLE: “LABS FOR” SLE
While SLE classification relies on the 2019 ACR/EULAR and 2012 Systemic Lupus International Collaborating Clinics (SLICC) criteria, dermatologists need a practical, streamlined approach to systematic screening in patients with CLE. The SLICC criteria, often favored in dermatology, incorporate cutaneous features such as oral ulcers, nonscarring alopecia, and discoid lupus lesions, alongside key autoantibodies (ANA, anti-dsDNA, anti-Sm).7,8 However, diagnostic delays of up to three years remain common, highlighting the need for simplified, real-world screening tools to facilitate earlier detection.9
To ensure a structured and efficient assessment, we propose the “LABS FOR” SLE mnemonic as a quick reference for essential SLE workup components in patients with CLE (Figure 1):
- Low complements (C3/C4)
- ANA with reflex
- Blood counts (complete blood count [CBC])
- Serologies (dsDNA, antiphospholipid antibodies [APLA], others if ANA+)
- Fatigue, fever, full ROS
- Organ function: complete metabolic panel (CMP)
- Renal check: urinalysis (UA), creatinine (Cr)
This high-yield checklist prompts dermatologists to routinely assess hallmark serologic and laboratory abnormalities, reducing the risk of missed systemic involvement. While SLE is heterogeneous, incorporating this mnemonic into dermatology practice can help expedite diagnosis, facilitate timely rheumatologic referral, and improve patient outcomes.
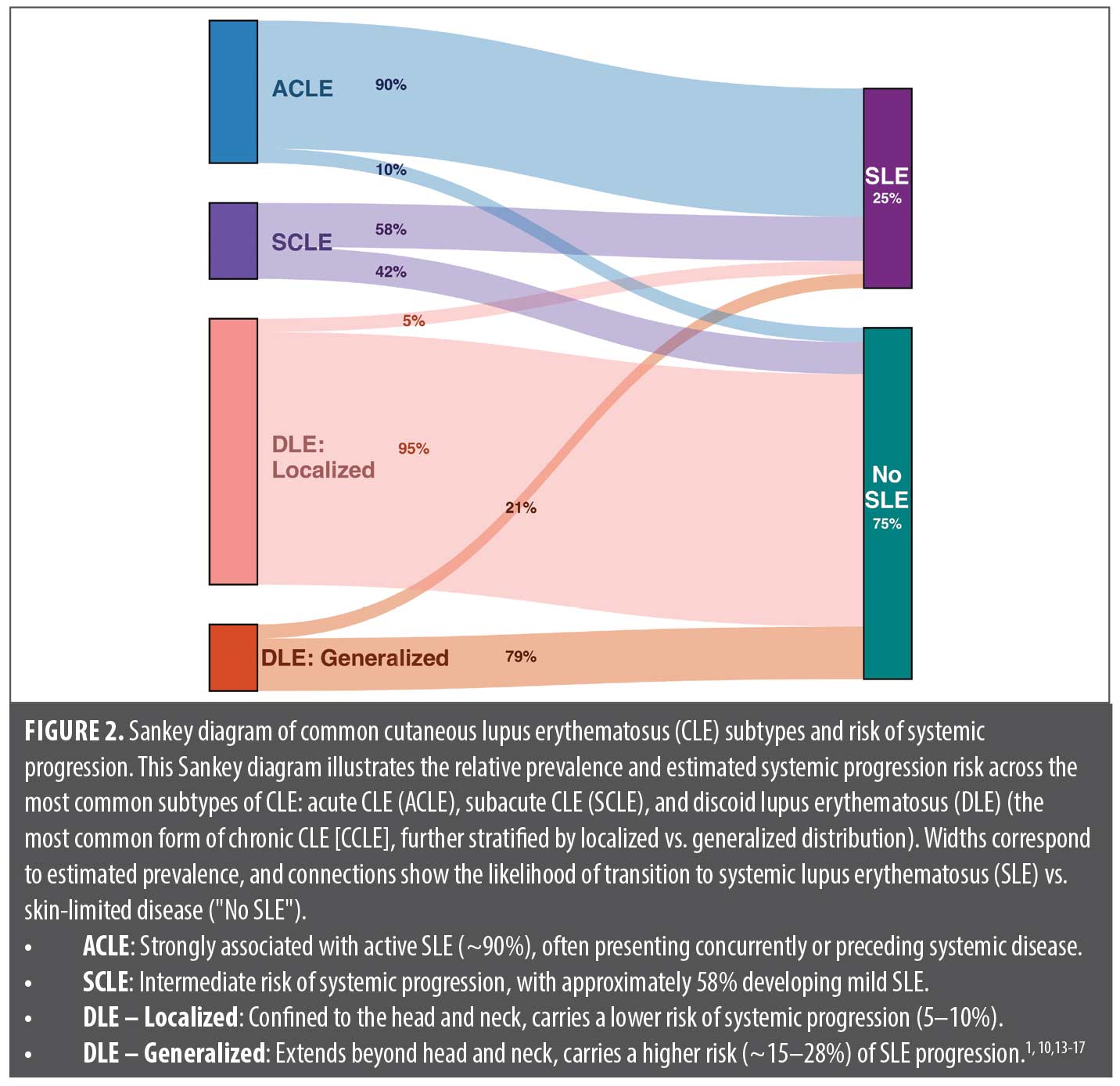
CLE Subtypes and SLE Risk
CLE encompasses a spectrum of disease, ranging from skin-limited forms to those closely associated with SLE. CLE is traditionally classified into acute (ACLE), subacute (SCLE), and chronic (CCLE) subtypes, though overlapping features occur in up to 30% of cases.1,10 Discoid lupus erythematosus (DLE) is the most common subtype of CCLE, representing at least half of all cases.1 These subtypes differ significantly in their likelihood of systemic involvement: ACLE is highly correlated with active SLE, SCLE carries a moderate risk, and most CCLE subtypes remain skin-limited, though exceptions exist.1,11,12 While we review previously reported rates of systemic progression across CLE subtypes, it is important to note that these estimates are largely based on the 1997 ACR classification criteria, which may overestimate SLE diagnosis in certain subtypes, such as SCLE, compared to 2019 ACR/EULAR criteria.6 Figure 2 illustrates the relative prevalence and systemic risk across key CLE subtypes. Recognizing these distinctions is essential for risk stratification, guiding treatment, and determining when systemic evaluation is warranted.
Acute Cutaneous Lupus Erythematosus (ACLE)
ACLE is the prototypical “lupus rash,” occurring during systemic lupus flares. The classic presentation is a transient, photosensitive malar rash sparing the nasolabial folds, though a more widespread morbilliform or papular eruption can appear on sun-exposed areas.12, 18 Unlike DLE, ACLE does not scar but may leave postinflammatory dyspigmentation.19 A severe toxic epidermal necrolysis (TEN)-like variant, characterized by diffuse blistering and epidermal detachment, signals high-risk SLE and must be distinguished from drug-induced TEN.20
ACLE is a strong indicator of systemic lupus; 30 to 50% of patients with SLE develop ACLE, and its presence warrants prompt systemic evaluation for nephritis, serositis, and hematologic involvement.1 Given its close association with active SLE, patients with ACLE often require rheumatologic comanagement.
Subacute Cutaneous Lupus Erythematosus (SCLE)
SCLE presents as a chronic, photosensitive, nonscarring eruption in annular-polycyclic or papulosquamous (psoriasiform) forms. Lesions favor sun-exposed areas but typically spare the face. Histologically, SCLE features interface dermatitis, and direct immunofluorescence often reveals a positive lupus band.1 Drug-induced SCLE should be considered in any patient presenting with these features. Other less common variants include vesiculobullous, erythrodermic, and Rowell’s syndrome (erythema multiforme-like) SCLE.21
Approximately 20 to 40% of SCLE cases are drug-induced, commonly triggered by thiazides, terbinafine, and tumor necrosis factor (TNF) inhibitors. Drug-induced SCLE is clinically indistinguishable from idiopathic SCLE, making serologic testing crucial, because anti-histone antibodies are positive in 90 to 95% of drug-induced cases, though clinical and laboratory profiles vary by drug.22
SCLE confers an intermediate systemic risk, with 50 to 60% of patients historically meeting SLE criteria; however, this estimate is largely derived from studies using the 1997 ACR criteria, under which patients with SCLE more readily qualified than under the 2019 ACR/EULAR criteria. Notably, even among patients with SCLE who meet criteria for SLE, major organ involvement is rare.1,23 Anti-Sjögren’s syndrome-related antigen A (SSA), or anti-Ro, is the hallmark serologic marker, present in approximately 70% of cases, though a subset (~5%) are ANA-negative but SSA-positive, known as “ANA-negative lupus.” Given this variability, dermatologists should screen patients with SCLE for systemic involvement with baseline serologies and ongoing monitoring. Screening for SSA is particularly important when clinical suspicion is high or concern for risk of neonatal lupus is present.24,25
Chronic Cutaneous Lupus Erythematosus (CCLE)
DLE is the most common CCLE subtype and presents as scarring, hyperkeratotic plaques with follicular plugging, often affecting the face, ears, and scalp.26 Systemic progression risk varies significantly by disease distribution. Localized DLE, confined to the head and neck, carries a relatively low risk of developing SLE, typically estimated between 5 to 10%. In contrast, generalized DLE, defined by involvement of sites below the neck (eg, trunk or extremities), is associated with markedly higher risk reported in the range of 15 to 28%.17 Risk of systemic progression increases with positive ANA, dsDNA, or hypocomplementemia.27 Of note, per the Brigham Lupus Registry, when systemic conversion occurs, it typically does so within 1 to 2 years.28
Notably, DLE itself contributes four points in the ACR/EULAR SLE classification system, meaning some patients with DLE and serologic abnormalities may meet SLE criteria despite minimal systemic symptoms.6 Further, compared to SLE without DLE, SLE with DLE is associated with a lower risk of arthritis and serositis but carries a similar risk of nephritis, end-stage renal disease (ESRD), and other major organ involvement.29
Given this heterogeneity, dermatologists should distinguish localized versus generalized DLE, as generalized cases warrant closer systemic surveillance. Baseline serologies and periodic monitoring are essential, particularly in patients with evolving systemic symptoms. Aggressive early treatment prevents disfigurement, while ongoing surveillance ensures timely recognition of systemic progression.
Summary of Common CLE Subtypes and SLE Risk
The risk of SLE varies significantly across CLE subtypes, necessitating a stratified approach to evaluation. ACLE (eg, malar rash) is highly predictive of SLE, often occurring concurrently with systemic disease. SCLE carries a moderate risk (~50%), with most cases involving mild systemic disease rather than severe organ involvement. CCLE has a more variable risk profile.1 Given this heterogeneity, all patients with CLE warrant baseline serologic screening, and some may harbor serologic or subclinical systemic disease despite an otherwise localized presentation. Identifying clinical, serologic, and genetic markers of progression remains critical to optimizing long-term monitoring and management.
Risk Factors for CLE Progression to Systemic Disease
While the majority of patients with CLE do not develop SLE, identifying those at higher risk remains a critical area of research. Studies have highlighted clinical, demographic, serologic, and genetic factors that predict progression, allowing dermatologists to stratify patients for appropriate monitoring and referral. Those with high-risk features may require closer surveillance and early rheumatology involvement, while patients lacking systemic risk markers may be followed with routine dermatologic care. Below, we summarize the latest evidence on predictors of CLE-to-SLE progression.
Serologic markers. Autoantibodies play a pivotal role in predicting systemic progression in CLE. A positive ANA is a key risk factor; studies consistently show that patients with CLE who develop SLE are more likely to have had a positive ANA at baseline.30 The titer also matters—a recent Etudes des Maladies Systémiques en Dermatologie (EMSED) registry study (2023) of 164 patients with DLE identified ANA ≥1:320 as a strong independent predictor of severe SLE (odds ratio [OR]: ~15).30 Similarly, high-titer ANA was linked to SLE progression in a 2012 analysis by Chong et al.27 In healthy individuals, ANA positivity is seen in around 31% at 1:40 dilution, 13% at 1:80, 5% at 1:160, and only 3% at 1:320, highlighting the need to interpret ANA in clinical context rather than as a standalone marker.31
Beyond ANA, anti-dsDNA, and anti-Sm antibodies strongly suggest occult or impending SLE, particularly in patients with DLE, where dsDNA positivity often correlates with future lupus nephritis.32 Anti-Ro/SSA, common in SCLE, does not always indicate systemic disease but warrants monitoring for SLE or Sjögren’s syndrome. Low complement levels (C3, C4) are another red flag, as hypocomplementemia is rare in isolated CLE but a hallmark of systemic lupus activity.1
Emerging biomarkers like the AVISE® Connective Tissue Disease (CTD) test, which detects complement activation products (EC4d, BC4d), may further refine risk stratification. One study found that 65% of AVISE-positive patients (without prior SLE) later developed systemic disease, compared to only 10% of AVISE-negative patients.33 While promising, such tests remain adjunctive and require further validation.
Clinical and demographic factors. Certain patient characteristics and cutaneous features can help stratify the risk of systemic progression. Younger age at CLE onset is a key factor, as patients diagnosed with DLE before 25 years of age have nearly threefold higher odds of developing SLE. Race and skin phototype also play a role, with patients of color (ie, Fitzpatrick skin type V/VI) exhibiting a 2.7-fold increased risk of progression, likely due to genetic predisposition and healthcare disparities.30
Disease extent and distribution are critical prognostic markers. Generalized DLE carries a significantly higher SLE risk than localized disease, with multiple anatomic sites and refractory skin lesions warranting closer systemic evaluation. Certain mucocutaneous findings, such as periungual telangiectasias, Raynaud’s phenomenon, extensive oral ulceration, and nailfold capillary changes, may signal underlying CTD. Arthralgias, even if subclinical, and unexplained severe fatigue should also raise suspicion.27 While SLE is more common in women, CLE in men may be more likely to progress to systemic disease, potentially due to a higher immune threshold for autoimmunity. Lastly, smoking, while strongly linked to CLE flares, may paradoxically correlate with a lower risk of systemic involvement; however, cessation remains critical for disease control.30
Genetic predisposition. Genetic factors influence the likelihood of systemic progression. The HLA-B8/DR3 haplotype is strongly linked to SCLE and anti-Ro positivity, often predisposing patients to photosensitive lupus with autoantibodies but without severe systemic disease.34,35 Meanwhile, HLA-DR2, HLA-DR4, and complement deficiencies (C1q, C2, C4) are associated with a higher likelihood of SLE, with complete complement deficiencies almost invariably leading to SLE.34 Integrin Subunit Alpha M (ITGAM) polymorphisms, which impact C3 function, have been linked specifically to DLE, reinforcing the idea that some genetic variants preferentially drive skin-predominant disease.36 Further, high expression of chemokine CXCL10 may indicate more systemic involvement.37
Though genetic testing is not routine in CLE, family history provides valuable insight. Patients with multiple relatives affected by SLE or other autoimmune diseases may carry a higher genetic load for systemic disease. The type I interferon (IFN) pathway is a key immunogenetic driver, with high systemic IFN activity (“IFN signature”) correlating with greater risk of SLE transition, while a skin-restricted IFN response may indicate skin-limited lupus.38 In the future, gene expression profiling or polygenic risk scores may refine risk stratification, but for now, clinical factors such as young age and non-White race often reflect an underlying genetic predisposition toward systemic autoimmunity.
Updated ACR/EULAR Classification Criteria for SLE: Implications for Dermatology.
The 2019 ACR/EULAR criteria redefine SLE classification with a mandatory ANA ≥1:80 as an entry requirement, effectively excluding ANA-negative lupus. This has direct implications for dermatologists, particularly in SCLE, where SSA/Ro-positive, ANA-negative cases occur. Separate SSA/Ro testing remains appropriate in clinically suspected cases.1
After meeting the ANA criterion, patients must accumulate 10 or more points across weighted clinical and immunologic domains. CLE is consolidated under a single “mucocutaneous” domain, with ACLE (six points) carrying the highest weight, followed by SCLE or DLE (four points), and oral ulcers or nonscarring alopecia (two points each). Only the highest-scoring cutaneous feature is counted.6
For dermatologists, these criteria help differentiate CLE-limited cases from those warranting systemic evaluation. A patient who has ANA-positive DLE (four points) with arthritis (six points) qualifies as SLE (10 points), while one with only low complements (four points) does not (eight points). Notably, fewer patients with CLE qualify for SLE under ACR/EULAR than under the 2012 SLICC criteria, which permit ANA-negative cases and count acute and chronic CLE separately.39, 40 Thus, some patients with CLE who fail ACR/EULAR may still be classified as SLE under SLICC, and vice versa.
These updates reinforce the need for SLE screening in CLE, appropriate ANA testing, and informed triage to rheumatology.
Screening and Monitoring Patients with CLE for Systemic Involvement
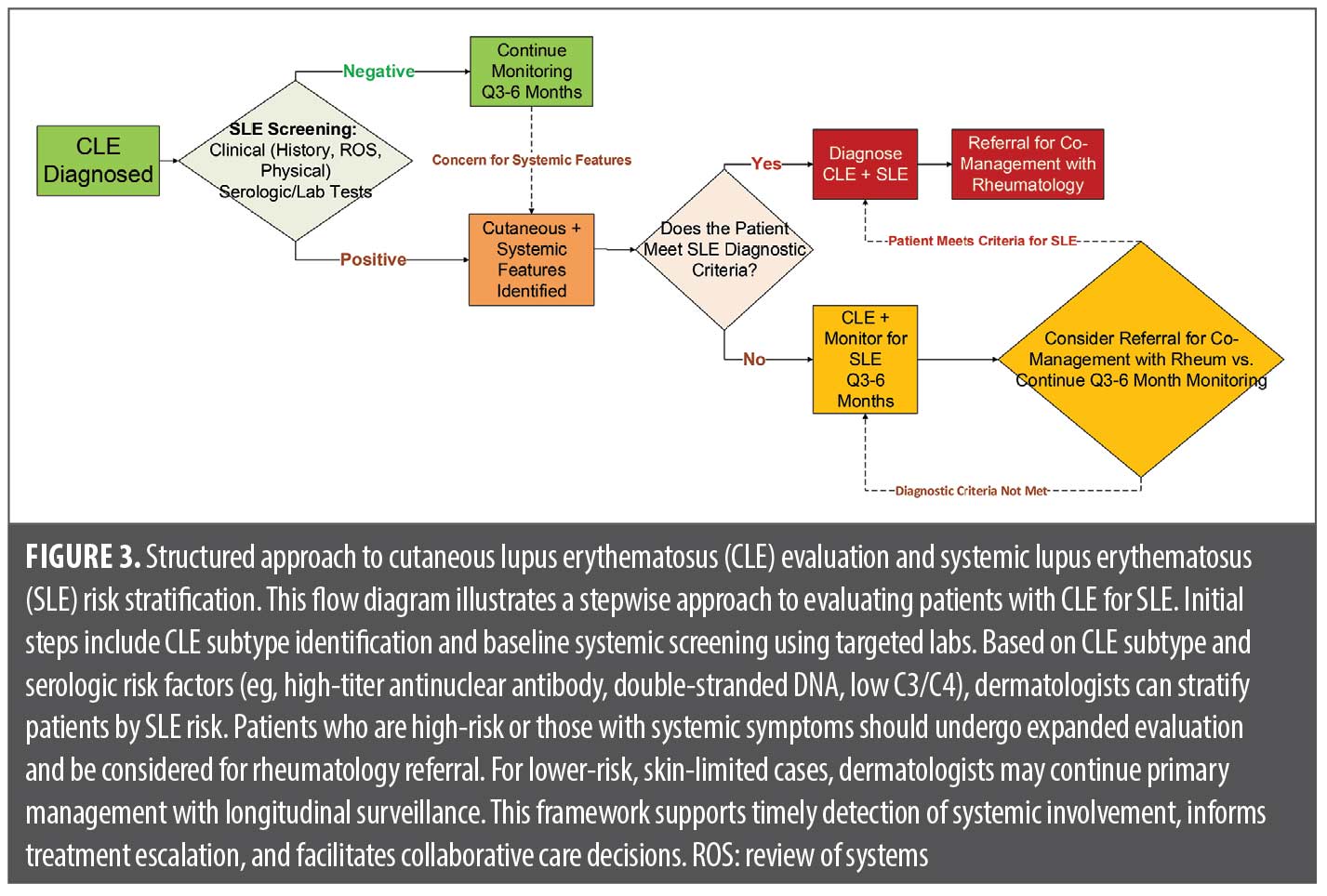
A systematic approach to screening and surveillance is critical for detecting systemic involvement in CLE. Baseline evaluation at diagnosis, followed by regular follow-up every 3 to 6 months, helps identify high-risk patients while minimizing unnecessary testing in those with skin-limited disease. Key assessments include serologic and laboratory markers, clinical symptom review, and targeted follow-up based on risk stratification.41 Of note, ANA testing should be performed by indirect immunofluorescence (IIF), which remains the standard per the ACR due to superior sensitivity and nuanced pattern recognition. Solid-phase assays may miss clinically significant antibodies, particularly in early or cutaneous-limited disease. Additionally, if positive, reflex testing to relevant extractable nuclear antigens (ENA) is preferred over blanket ENA panels, which may be costly and clinically ambiguous.42,43 If systemic features emerge, including constitutional symptoms, arthritis, cytopenias, or renal abnormalities, prompt referral to rheumatology is advised (Figure 3).41
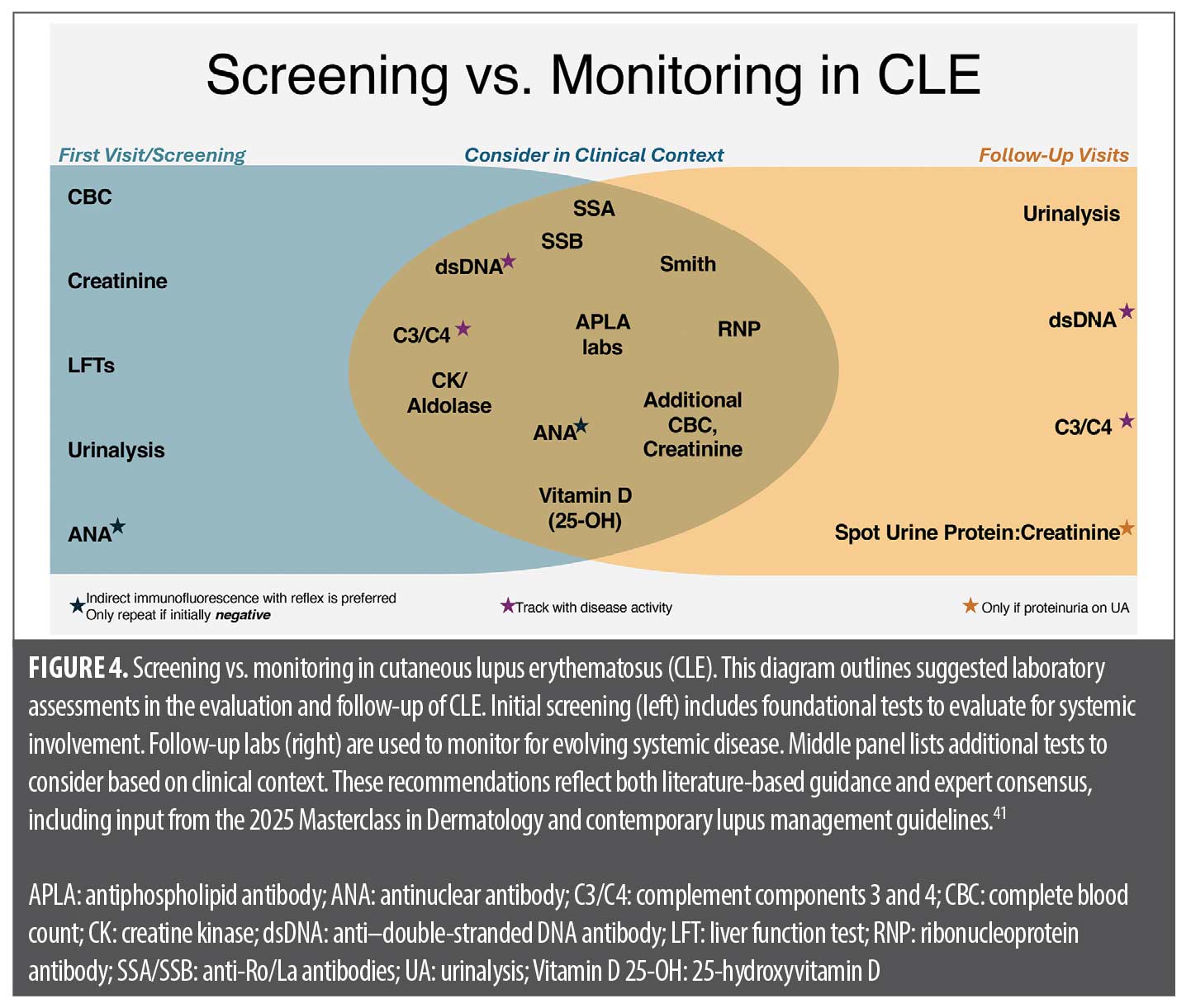
Initial laboratory work-up (baseline screening). All newly diagnosed patients with CLE should undergo a targeted laboratory panel to assess systemic involvement.41 This includes:
- CBC: Screens for cytopenias (anemia, leukopenia, thrombocytopenia)
- Serum creatinine and liver function tests (LFTs): Evaluates renal function and establishes a baseline for potential medication effects
- Urinalysis (UA): Essential for detecting proteinuria or hematuria, which may indicate occult nephritis. Any abnormality warrants further quantification (protein/creatinine ratio) and nephrology referral.
- ANA Test: IIF (Hep-2 cells) is the gold standard. A high titer (≥1:160) raises SLE suspicion, but even a titer of 1:80 is significant in a patient with CLE. If ANA is negative, test anti-Ro/SSA separately in suspected or higher-risk SCLE cases (eg, in pregnancy due to concern for neonatal lupus) given the rare possibility of ANA-negative but SSA-positive SCLE.
Expanded work-up (if indicated). For patients with positive ANA, symptoms, or abnormal baseline labs, further autoantibody and complement testing should be considered:
- Autoantibody panel: Anti-dsDNA (renal risk), anti-Sm (high SLE specificity), anti–Ro/La antibodies (SCLE, neonatal lupus, Sjögren’s), anti-ribonucleoprotein antibody [anti-RNP] (overlap syndromes), anti-Histone (drug-induced), and APLA (clotting risk).22,25,44 Some experts advocate APLA testing in all patients with ANA+ CLE.
- Complement Levels (C3, C4): Low C3/C4 suggests active immune complex formation and greater systemic risk.
- Inflammatory markers (erythrocyte sedimentation rate [ESR], C-reactive protein [CRP]): ESR elevation is common in SLE flares, whereas CRP is typically normal unless infection or serositis is present.
- Others as indicated: If muscle pain or weakness is present, creatine kinase (CK) and aldolase might be checked to rule out myositis overlap. If urine showed protein, a nephrology consult might be next.
Patients with CLE do not universally require second-line tests, but a comprehensive serologic panel is reasonable at baseline, even if asymptomatic, to avoid missing early systemic disease. Follow-up surveillance should focus on new symptoms, worsening skin disease, or abnormal labs, prompting repeat testing or rheumatology referral when needed.
Follow-up and monitoring. Once baseline evaluation is complete, ongoing surveillance should be tailored based on disease activity and risk. Patients with active CLE should be seen every 3 to 6 months, while those with stable disease may require only annual follow-ups. Each visit should include interval history for new systemic symptoms and a targeted exam (skin, joints, lymph nodes). Laboratory monitoring should be strategic, focusing on tests that provide meaningful information:
- ANA does not require repetition if positive initially. While ANA titers fluctuate, a previously positive result rarely normalizes, and retesting does not change management.
- UA is a key periodic test, recommended every 6 to 12 months in patients with CLE to detect silent nephritis. If proteinuria was previously noted, a protein/creatinine ratio should be monitored.
- CBC and metabolic panel should be repeated every 6 to 12 months or more frequently if systemic medications (eg, methotrexate, antimalarials) are being used.
- Anti-dsDNA and complement levels (C3, C4) track disease activity and should be checked periodically or if new symptoms arise (rash flare, arthralgia, etc.). Some dermatologists repeat them annually in patients with CLE-only as a precaution.45
- Emerging tests (eg, AVISE® CTD panel) measuring C4d activation products may help predict systemic progression, though these are still evolving.
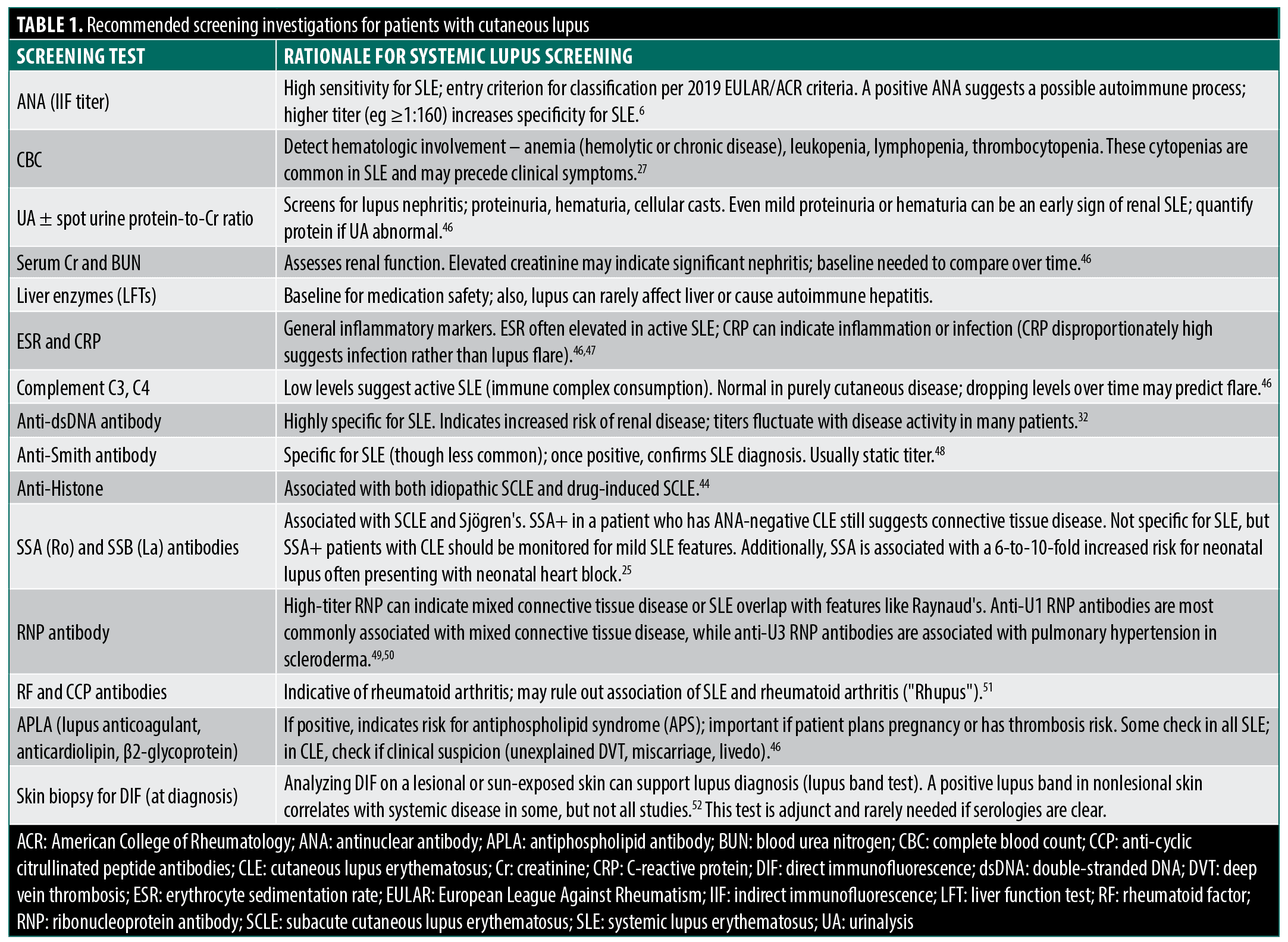
Patients with CLE should be counseled on symptoms warranting urgent evaluation, including persistent fever, unexplained weight loss, worsening fatigue, new joint pain/swelling, pleuritic chest pain, edema or foamy urine (renal involvement), and neurological symptoms (headaches, confusion, seizure-like activity). Dermatologists should also monitor for cutaneous changes that may signal systemic transition, such as a patient with DLE developing an acute malar rash or vasculitic lesions. Recommended screens in initial visits versus ongoing monitoring are illustrated in Figure 4. Table 1 summarizes recommended baseline screening tests for patients with CLE and their rationale.
Collaborative Management Between Dermatology and Rheumatology
Effective lupus care requires close collaboration between dermatologists and rheumatologists, as neither specialty alone addresses the full disease spectrum. Early collaboration ensures a cohesive treatment strategy, balancing cutaneous and systemic disease control while minimizing medication redundancies. Therapeutic decisions should be tailored to both skin and systemic manifestations. Antimalarials remain first-line for CLE and SLE, but when systemic therapy escalation is needed, dermatologists and rheumatologists should coordinate second-line choices based on disease phenotype. While not exhaustive, the following examples of various symptoms observed in patients with CLE illustrate how therapeutic choices may be tailored to different clinical scenarios:
- Arthritis: Methotrexate or belimumab
- Nephritis: Mycophenolate mofetil (MMF) preferred over azathioprine; combination approaches may include MMF + belimumab, MMF + cyclosporine, MMF + rituximab
- Interstitial lung disease (ILD): MMF, possibly rituximab
- Antiphospholipid syndrome or hypercoagulability: Avoid or approach thalidomide with caution
- Refractory systemic lupus despite standard treatments: Belimumab or anifrolumab
- Moderate-to-severe active systemic disease: Avoid thalidomide monotherapy; combination therapy is preferred
Given the dual benefits of many immunosuppressants on both skin and systemic manifestations, treatment plans should be clearly communicated to avoid duplication and optimize monitoring. Alternating dermatology and rheumatology visits, sharing lab monitoring (eg, CBC, CMP, UA), and dividing long-term surveillance tasks (eg, dermatologists tracking hydroxychloroquine adherence and skin cancer risk while rheumatologists oversee bone health and vascular complications) improve efficiency and patient adherence. Consistent patient education on photoprotection, smoking cessation, and pregnancy planning further mitigates complications. In refractory cases, combined expertise can guide advanced strategies, such as integrating thalidomide for severe DLE alongside systemic immunosuppression or combining laser therapy for scarring with aggressive disease control. Emerging multidisciplinary rheumatology-dermatology clinics are poised to streamline care, improving outcomes through comprehensive, coordinated management.49
Conclusion
Lupus is a multisystem disease in which cutaneous manifestations are often an early sign, placing dermatologists at the forefront of diagnosis and management. The 2025 Masterclass in Dermatology reinforced the need for dermatologists to recognize CLE subtypes, assess SLE risk, and implement systematic screening and monitoring strategies. Understanding the variable likelihood of systemic progression given specific clinical presentations allows for tailored prognostic counseling and surveillance.
To support efficient evaluation in clinical practice and reinforce durable learning among trainees, we introduced the “LABS FOR” SLE mnemonic, which is a practical, memorable tool for guiding SLE screening in patients with cutaneous lupus. When used alongside the updated 2019 ACR/EULAR classification criteria, which improve diagnostic specificity, such tools facilitate timely recognition of systemic involvement.
Interdisciplinary collaboration remains key. As skin findings often mirror underlying immune dysregulation, coordinated care with rheumatology ensures that both cutaneous and systemic aspects of lupus are addressed. By applying evidence-based strategies and fostering strong comanaging partnerships, dermatologists can improve early diagnosis, disease control, and long-term outcomes in patients with lupus—not just treating the skin, but also safeguarding against systemic disease.
References:
- Stull C, Sprow G, Werth VP. Cutaneous involvement in systemic lupus erythematosus: a review for the rheumatologist. J Rheumatol. 2023;50(1):27-35.
- Grönhagen CM, Gunnarsson I, Svenugsson E, Nyberg F. Cutaneous manifestations and serological findings in 260 patients with systemic lupus erythematosus. Lupus. 2010;19(10):1187-1194.
- Zhang AJ, Ezeh N, Childs B, et al. Cutaneous lupus matters: independent and additive quality-of-life burden shown by 2022 WLFGI Survey. J Eur Acad Dermatol Venereol. 2025.
- Durosaro O, Davis M, Reed KB, Rohlinger AL. Incidence of cutaneous lupus erythematosus, 1965-2005: a population-based study. Arch Dermatol. 2009;145(3):249-253.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum.1997;40(9):1725.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78(9):1151-1159.
- Goldman N, Han J, LaChance A. Diagnosis and management of cutaneous manifestations of autoimmune connective tissue diseases. Clin Cosmet Investig Dermatol. 2022;15:2285-2312.
- Aringer M, Leuchten N, Johnson SR. New criteria for lupus. Curr Rheumatol Rep. 2020;22(6):18.
- Sanchez‑Rodriguez A, Meade‑Aguilar JA, Yang JX, et al. Clinical presentation, care pathways, and delays in access to specialized care in patients with systemic lupus erythematosus: a study from Lupus Midwest Network (LUMEN). Arthritis Care Res (Hoboken). 2025;77(4):451-459.
- Biazar C, Sigges J, Patsinakidis N, et al. Cutaneous lupus erythematosus: first multicenter database analysis of 1002 patients from the European Society of Cutaneous Lupus Erythematosus (EUSCLE). Autoimmun Rev. 2013;12(3):444‑454.
- Hocaoğlu M, Davis MDP, Osei‑Onomah SA, et al. Epidemiology of cutaneous lupus erythematosus among adults over four decades (1976‑2018): a Lupus Midwest Network (LUMEN) study. Mayo Clin Proc. 2022;97(12):2282-2290.
- Vale E, Garcia LC. Cutaneous lupus erythematosus: a review of etiopathogenic, clinical, diagnostic and therapeutic aspects. An Bras Dermatol. 2023;98(3):355-372.
- O’Brien JC, Chong BF. Not just skin deep: systemic disease involvement in patients with cutaneous lupus. J Investig Dermatol Symp Proc. 2017;18(2):S69-S74.
- Klein RS, Morganroth PA, Werth VP. Cutaneous lupus and the Cutaneous Lupus Erythematosus Disease Area and Severity Index instrument. Rheum Dis Clin North Am. 2010;36(1):33–51.
- Watanabe T, Tsuchida T. Classification of lupus erythematosus based upon cutaneous manifestations: dermatological, systemic and laboratory findings in 191 patients. Dermatology. 1995;190(4):277–283.
- Vera-Recabarren MA, García-Carrasco M, Ramos-Casals M, et al. Comparative analysis of subacute cutaneous lupus erythematosus and chronic cutaneous lupus erythematosus: clinical and immunological study of 270 patients. Br J Dermatol. 2010;162(1):91–101.
- Merola, J.F., Overview of cutaneous lupus erythematosus, in UpToDate, D.S. Pisetsky, et al., Editors. 2025, Wolters Kluwer.
- Blake SC, Daniel BS. Cutaneous lupus erythematosus: a review of the literature. Int J Womens Dermatol. 2019;5(5):320-329.
- Cooper EE, Pisano CE, Shapiro SC. Cutaneous manifestations of “lupus”: systemic lupus erythematosus and beyond. Int J Rheumatol. 2021;2021:6610509.
- Romero LS, Bari O, Smith CJF, Schneider JA, Cohen PR. Toxic epidermal necrolysis-like acute cutaneous lupus erythematosus: report of a case and review of the literature. Dermatol Online J. 2018;24(5).
- Lee HJ, Sinha AA. Cutaneous lupus erythematosus: understanding of clinical features, genetic basis, and pathobiology of disease guides therapeutic strategies. Autoimmunity. 2006;39(6):433-444.
- Merola JF. Drug-induced lupus erythematosus. In: Lippincott WW, ed. The Clinical Management of Systemic Lupus Erythematosus. Schur PH, Massarrotti E, eds. Lippincott; 1996.
- Zhou W, Wu H, Zhao M, Lu Q. New insights into the progression from cutaneous lupus to systemic lupus erythematosus. Expert Rev Clin Immunol. 2020;16(8):829-837.
- Provost TT, Reichlin M. Antinuclear antibody-negative systemic lupus erythematosus. I. Anti-Ro(SSA) and anti-La(SSB) antibodies. J Am Acad Dermatol. 1981;4(1):84-89.
- Izmirly PM, Llanos C, Lee LA, et al. Cutaneous manifestations of neonatal lupus and risk of subsequent congenital heart block. Arthritis Rheum. 2010;62(4):1153-1157.
- Grönhagen CM, Fored CM, Granath F, Nyberg F. Cutaneous lupus erythematosus and the association with systemic lupus erythematosus: a population-based cohort of 1088 patients in Sweden. Br J Dermatol. 2011;164(6):1335–1341.
- Chong BF, Song J, Olsen NJ. Determining risk factors for developing systemic lupus erythematosus in patients with discoid lupus erythematosus. Br J Dermatol. 2012;166(1):29-35.
- Elman SA, Joyce C, Costenbader KH, Merola JF. Time to progression from discoid lupus erythematosus to systemic lupus erythematosus: a retrospective cohort study. Clin Exp Dermatol. 2020;45(1):89-91.
- Merola JF, Prystowsky SD, Iversen C, et al. Association of discoid lupus erythematosus with other clinical manifestations among patients with systemic lupus erythematosus. J Am Acad Dermatol. 2013;69(1):19-24.
- Fredeau L, Courvoisier DS, Ait Mehdi R, et al. Risk factors of progression from discoid lupus to severe systemic lupus erythematosus: a registry-based cohort study of 164 patients. J Am Acad Dermatol. 2023;88(3):551-559.
- Tan EM, Feltkamp TE, Smolen JS, et al. Range of antinuclear antibodies in “healthy” individuals. Arthritis Rheum. 1997;40(9):1601-1611.
- Yung S, Chan TM. Anti-dsDNA antibodies and resident renal cells – their putative roles in pathogenesis of renal lesions in lupus nephritis. Clin Immunol. 2017;185:40-50.
- Liang E, Taylor M, McMahon M. Utility of the AVISE Connective Tissue Disease test in predicting lupus diagnosis and progression. Lupus Sci Med. 2020;7(1):e000345.
- Graham RR, Ortmann W, Rodine P, et al. Specific combinations of HLA-DR2 and DR3 class II haplotypes contribute graded risk for disease susceptibility and autoantibodies in human SLE. Eur J Hum Genet. 2007;15(8):823-830.
- Chen HW, Barber G, Chong BF. The genetic landscape of cutaneous lupus erythematosus. Front Med (Lausanne). 2022;9:916011.
- Jarvinen TM, Hellquist A, Koskenmies S, et al. Polymorphisms of the ITGAM gene confer higher risk of discoid cutaneous than of systemic lupus erythematosus. PLoS One. 2010;5(12):e14212.
- Zhu JL, O’Brien JC, Barber G, Saxena R, Chong BF, et al. Elevated serum levels of C-X-C motif chemokine ligand 10 can distinguish systemic lupus erythematosus patients from cutaneous lupus erythematosus patients. J Am Acad Dermatol. 2021;85(4):1051-1054.
- Postal M, Vivaldo JF, Fernandez-Ruiz R, Paredes JL, Appenzeller S, Niewold TB, et al. Type I interferon in the pathogenesis of systemic lupus erythematosus. Curr Opin Immunol. 2020;67:87-94.
- Tarazi M, Gaffney RG, Kushner CJ, et al. Cutaneous lupus erythematosus patients with a negative antinuclear antibody meeting the American College of Rheumatology and/or Systemic Lupus International Collaborating Clinics criteria for systemic lupus erythematosus. Arthritis Care Res (Hoboken). 2019;71(11):1404-1409.
- Black SM, Walocko F, Li X, Chong BF. Development of systemic lupus in patients with cutaneous lupus using the 2012 Systemic Lupus International Collaborating Clinics (SLICC) classification criteria for systemic lupus erythematosus. J Am Acad Dermatol. 2021;85(1):200–202.
- Lu Q, Long H, Chow S, et al. Guideline for the diagnosis, treatment and long-term management of cutaneous lupus erythematosus. J Autoimmun. 2021;123:102707.
- Meroni PL, Schur PH. ANA screening: an old test with new recommendations. Ann Rheum Dis. 2010;69(8):1420-1422.
- Tonutti E, Bizzaro N, Morozzi G, et al. The ANA-reflex test as a model for improving clinical appropriateness in autoimmune diagnostics. Auto Immun Highlights. 2016;7(1):9.
- Merola JF. Drug-induced lupus. In: Pisetsky DS, et al., editors. UpToDate. Wolters Kluwer; 2025.
- Xie L, Lopes Almeida Gomes L, Stone CJ, et al. An update on clinical trials for cutaneous lupus erythematosus. J Dermatol. 2024;51(7):885-894.
- Tunnicliffe DJ, Singh-Grewal D, Kim S, et al. Diagnosis, Monitoring, and Treatment of Systemic Lupus Erythematosus: A Systematic Review of Clinical Practice Guidelines. Arthritis Care Res (Hoboken). 2015;67(10):1440-52.
- Littlejohn E, Marder W, Lewis E, et al. The ratio of erythrocyte sedimentation rate to C-reactive protein is useful in distinguishing infection from flare in systemic lupus erythematosus patients presenting with fever. Lupus. 2018;27(7):1123-9.
- van Beers J, Schreurs MWJ. Anti-Sm antibodies in the classification criteria of systemic lupus erythematosus. J Transl Autoimmun. 2022;5:100155.
- Chevalier K, Chasagnon G, Leonard-Louis S, et al.. Anti-U1RNP antibodies are associated with a distinct clinical phenotype and worse survival in patients with systemic sclerosis. J Autoimmun. 2024;146:103220.
- Aggarwal R, Lucas M, Fertig N, et al. Anti-U3 RNP autoantibodies in systemic sclerosis. Arthritis Rheum. 2009;60(4):1112-1118.
- Chen X, Li Y, Chen X, et al. Rhupus syndrome: a unique disease overlapping systemic lupus erythematosus and rheumatoid arthritis. Arch Dermatol Res. 2024;317(1):127.
- Wongtada C, Kerr SJ, Rerknimitr P. Lupus band test for diagnostic evaluation in systemic lupus erythematosus. Lupus. 2022;31(3):363-366.
- Samycia M, McCourt C, Shojania K, Au S, et al. Experiences from a combined dermatology and rheumatology clinic: a retrospective review. J Cutan Med Surg. 2016;20(5):486-489.
Optimizing Surgical Tray Setup and Instrument Selection in Dermatologic Surgery