
J Clin Aesthet Dermatol 2018;11(10):S5–S23
A Message from the Guest Editor and MauiDerm Program Director
Dear Colleagues:
Each year, Maui Derm strives to bring to the podium the most up-to-date and cutting edge scientific and clinical developments in the field of dermatology. Maui Derm 2018 was no exception—our outstanding faculty delivered a mind-numbing amount of clinical information in multiple areas of dermatology. One therapeutic area in particular—psoriasis—has demonstrated a rapid expansion of new therapeutic options. In this supplement to The Journal of Clinical and Aesthetic Dermatology (JCAD), we’ve captured a select group of Maui Derm 2018 presentations we feel represents the key recent clinical developments in psoriasis. Here, we provide information on newly discovered mechanisms of action, emerging and newly approved drugs, and broadening therapeutic indications for existing drugs in the field of psoriasis.
We all understand the importance of staying up to date on the latest clinical developments in dermatology so that we can provide our patients with the best care possible. As busy dermatologists, however, it isn’t always possible to attend educational events such as Maui Derm. We hope the information presented in this supplement assists clinicians in determining how best to prescribe these emerging therapies as they come to market, in order to achieve optimal treatment outcomes in patients.
We hope you can join us January 26–30, 2019, in Maui next year for what promises to be another outstanding educational event. If your schedule prevents you from joining us in 2019, however, we hope to once again provide you with highlights from some of the key presentations from our meeting faculty.
With aloha,
George Martin, MD
Dermatology Associates, Khei, Maui, Hawaii; MauiDerm 2018 Program Director
Presenters:
Andrew Blauvelt, MD, MBA
Oregon Medical Research Center, Portland, Oregon
Joel M. Gelfand, MD, MSCE FAAD
University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania
Linda Stein Gold, MD
Henry Ford Health System, Detroit, Michigan
Arthur Kavanaugh, MD
Professor of Medicine and Director of the Center for Innovative Therapy (CIT) at UC San Diego, California
Craig L. Leonardi, MD
Associate Clinical Professor of Dermatology, St. Louis University Medical School, St. Louis, Missouri
Bruce E. Strober, MD, PhD
Professor and Chair, University of Connecticut Health Center, Department of Dermatology, Farmington, Connecticut
Updates in Psoriasis Management
Based on selected presentations from Maui Derm 2018, held January 28–February 1, 2018, in Maui, Hawaii
by Jo Ann LeQuang
Ms. Lequang is Owner of LeQ Medical in Angleton, Texas, Director of Scientific Communications at NEMA Research, Inc., in Naples, Florida, and Founding Director of No Baby Blisters in Colorado Springs, Colorado.
As more research is done in the pathogenesis of psoriasis and more potential targets are identified, new drugs and treatments develop that are changing the entire paradigm of care for patients with psoriasis. This year’s Maui Derm scientific sessions brought together key opinion leaders in the world of dermatology to discuss all aspects of dermatology with an emphasis on groundbreaking new science, novel therapeutic options, and insights into optimal care. This article is based on the presentations of Dr. Bruce E. Strober, who presented the latest key data from 2017; Dr. Joel M. Gelfand, who updated conference participants on new findings in psoriasis comorbidities and how these finding could—and should—impact clinical practice; Dr. Andrew Blauvelt, who provided an update on new findings in psoriasis pathogenesis and genetic involvement; Dr. Arthur Kavanaugh, who reported on psoriatic arthritis with an emphasis on new treatments; Dr. Craig Leonardi, who discussed the latest biologic products in the pipeline and on the market; and Dr. Linda Stein Gold, who addressed current topical treatment, misunderstandings about topical care, and the role of “ designer vehicles.”
Psoriasis: New Data from 2017
Based on the presentation by Bruce E. Strober, MD, PhD, Professor and Chair, University of Connecticut Health Center, Department of Dermatology, Farmington, Connecticut
Four significant studies emerge when perusing the new data from 2017, all of which can help guide clinical care of patients with psoriasis. The first involves treating psoriasis in pregnant and lactating women. Treating psoriasis in any special population can pose clinical challenges, but new and encouraging answers are available for certolizumab pegol. Second, the prevalent and distressing condition of genital psoriasis has always been difficult for patients and prescribers. New 2017 data on ixekizumab as a potential course of treatment holds promise. Third, novel treatments for psoriasis seem to abound but the issue of durability of results is relevant to patients and prescribers. This year, long-term data on secukinumab durability are available to help guide clinical decision-making. Finally, the risk of non-melanoma skin cancer (NMSC) and other cutaneous malignancies has been further studied in order to better understand what contributory role biologics might play in their development.
Certolizumab pegol in pregnancy. Pregnant women represent an important special population in which inflammatory diseases can be particularly difficult to treat due to the risk of transference to the child in utero. CRIB was a prospective, multicenter, post-market, pharmacokinetic study that evaluated the placental transfer of certolizumab pegol (CZP).1 Sixteen pregnant women were entered into and completed the study. The patients had to be at least 30 weeks pregnant, with a diagnosis of an approved CZP indication—psoriatic arthritis, rheumatoid arthritis, Crohn’s disease, or ankylosing spondylitis—and receive the last dose of CZP 35 days or less before delivery. Blood samples for measurement of CZP plasma concentration were taken from mothers, umbilical cords, and neonates at delivery; blood was drawn again from the infants at four and eight weeks post-delivery. At delivery, maternal CZP plasma levels from the 16 patients fell within the anticipated therapeutic range (24.4µg/mL, range 5.0– 49.4). Sixteen neonates were included in the study, but two were excluded from per-protocol datasets, one because of missing data at birth, the other because of implausible pharmacokinetic data. Of the 14 neonates in the per-protocol study, there was no quantifiable level of plasma CZP in 13 infants (<0.032µg/mL) and a minimal level in one (0.042µg/mL). At Weeks 4 and 8, none of the infants had quantifiable plasma levels of CZP. Umbilical cord samples were taken from the 16 patients but one was excluded due to missing data; of the 15 included umbilical cords, three had quantifiable levels of CZP, the highest of which was 0.048µg/mL. Thus, the CRIB clinical trial demonstrated that there was little to no CZP transfer from mother to child and that there was no fetal exposure to CZP during the third trimester of pregnancy. These findings suggest that CZP can be safely administered to pregnant women, when appropriate, without the risk of placental transfer.
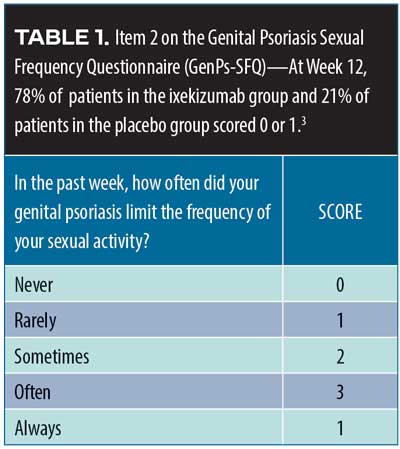
Ixekizumab in genital psoriasis. Genital psoriasis affects up to 63 percent of all patients with psoriaris2 and can be difficult to treat. A recent randomized, double-blind, placebo-controlled Phase IIIb study (IXORA-Q)3 evaluated the potential of ixekizumab for the treatment of genital psoriasis. A total of 149 patients with moderate-to-severe genital psoriasis with body surface area (BSA) greater than one percent were randomized to receive either placebo (n=74) or ixekizumab at recommended doses (n=75). Moderate-to-severe genital psoriasis was defined by the Physician’s Global Assessment of Genitalia (sPGA-G) as having a score of three or higher. The primary endpoints were patients who score zero or one on the sPGA-G, the overall sPGA score, a three-point or more improvement over baseline on the itching numeric rating scale, and response to Item 2 on the Genital Psoriasis Sexual Frequency Questionnaire (GenPs-SFQ), which asked if genital psoriasis limited sexual activity (Table 1). The ixekizumab group received the United States Food and Drug Administration (FDA)-approved psoriasis dosing of 160mg of ixekizumab at Week 0, then 80mg every two weeks through Week 12; the placebo group received placebo on the same schedule. After 12 weeks, patients entered an open-label treatment phase for a total of 52 weeks. Most of the IXORA-Q patients were male (75.8%). The mean age of patients was 43.7± 2.7 years and mean weight was 93.5±24.7kg. Among the participants, 52.3 percent had previously used non-biologic systemic therapies, and19.5 percent had previously used biologic systemic therapies to treat their psoriasis. By Week 12, significantly more patients in the ixekizumab group had clear or almost clear genital-area skin compared to placebo (73.3% vs. 8.1%, p<0.001). Fifty-six percent of ixekizumab patients had completely clear genital-area skin at 12 weeks compared to placebo (5.4%), p<0.001. Patients also reported at Week 12 a clinically meaningful relief from itchiness over baseline with ixekizumab versus placebo (59.7% vs. 8.3%, p<0.001), with a statistically significant difference emerging at Week 2. Item 2 on the GenPs-SFQ (Table 1) found that significantly more patients in the ixekizumab group reported that genital psoriasis never or rarely limited sexual activity at Week 12 versus placebo (78.4% vs. 21,4%, p<0.001) with the difference achieving statistical significance by Week 1 (21.6% vs. 4.8%, p=0.036). Treatment-emergent adverse events (TEAEs) were more frequent with ixekizumab than placebo; most were mild to moderate. The most commonly reported TEAEs with ixekizumab were respiratory tract infection, injection site reactions, oropharyngeal pain, pruritus, back pain, eczema, and hypertension. Severe TEAEs occurred more frequently among the placebo group than the ixekizumab group (4.1% placebo vs. 1.3% ixekizumab). Six patients discontinued the study (6.8% of placebo group and 1.3% of ixekizumab group) because of adverse effects.3
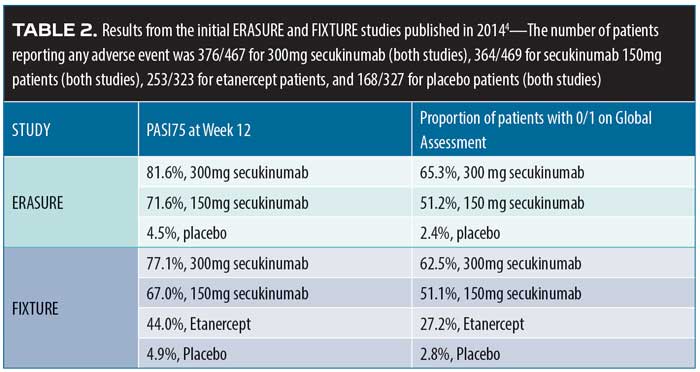
Secukinumab durability. In 2014, the results of two Phase III studies, Efficacy of Response and Safety of Two Fixed Secukinumab Regimens in Psoriasis (ERASURE, N=738) and Full-year Investigative Examination of Secukinumab vs. Etanercept Using Two Dosing Regimens to Determine Efficacy in Psoriasis (FIXTURE, N=1,306) were published.4 Patients with moderate-to-severe plaque psoriasis were recruited. Patients in the FIXTURE study were randomized to receive subcutaneous secukinumab 300mg or 100mg once a week for five weeks, then every four weeks thereafter; etanercept 50mg twice a week for 12 weeks, then weekly thereafter; or placebo. The study objective was to show the superiority of secukinumab over placebo at 12 weeks (defined as Psoriasis Area and Severity Index Score [PASI] of at least 75 percent over baseline [PASI75] and a score of “clear” or “almost clear,” that is, 0 or 1, on a five-point PGA). More patients in the secukinumab group at both doses achieved PASI75 at 12 weeks compared to patients in the placebo group or etanercept group ( Table 2).
The ERASURE and FIXTURE studies were then extended to evaluate long-term control of moderate-to-severe psoriasis following the withdrawal of secukinumab.4 PASI75 responders in the secukinumab 300mg and 150mg groups entered the study and were randomized to be treated for two years with secukinumab (same dose as original study) or placebo. Patients were monitored monthly and could not use any other psoriasis medications. Relapse in placebo patients, defined as greater than 50-percent loss of the previous gain in PASI score reduction, was treated by switching the patients to the secukinumab group. Among the placebo patients, 14 percent and 21 percent of the patients in the previous secukinumab 150mg and 300mg groups, respectively, did not relapse in the first year without medication; in the second year, six percent and 10 percent of the placebo patients who had previously been treated with secukinumab 150mg and 300mg, respectively, did not relapse. The meaning of these data is controversial and should not be interpreted to mean that the typical patient treated with secukinumab will achieve “remitted” control of his or her skin disease. In fact, without an active control group, it is impossible to know if this type of response is typical of many different therapies for psoriasis.
Risk of non-melanoma skin cancer (NMSC) with biologics versus methotrexate. The Psoriasis Longitudinal Assessment and Registry (PSOLAR) database was evaluated to determine the potential risk of NMSC in patients who used biologic therapy compared to those who were eligible for biologic treatment but took methotrexate instead.5 Collection of data was cut stopped on August 23, 2015, and was pooled from 12,090 patients representing 48,870 patient-years of follow-up. Within this group, 6,350 patients used biologics and 432 used methotrexate. Most of the patients in this registry study were Caucasian. This was a well-performed analysis with a comparator group. There were 72 new diagnoses of NMSC (48 basal cell carcinoma [BCC], 24 squamous cell carcinoma [SCC]). This signals a declining rate of BCC with ustekinumab and a trend toward increasing rates of SCC with TNF-inhibitors. That said, the risk of skin cancer with all biologic drugs remains very low, and targeted monitoring of high-risk patients remains the sensible approach.
Risk of malignancy with systemic psoriasis treatment. Using the PSOLAR data, a retrospective, nested case-control analysis6 was conducted on the risk of malignancy for individuals who received systemic psoriasis treatment; this analysis was based on 12,090 registry participants who were followed for 4.2 years, with a maximum follow-up period of 8.2 years. The analysis included patients with newly diagnosed malignancies (other than NMSC) and excluded those with a history of cancer (other than NMSC). Four randomly chosen control patients were matched with each case by age at index date (±6 months), sex, geographical region, and date of enrollment. For the purposes of this study, “exposure” was defined as having received a dose of the study therapy within one year of the index date and stratified by the duration of therapy into three discreet categories: greater than 0 and less than three months; three months to less than 12 months; and 12 months or longer. A total of 252 malignancy cases were found and matched to a total of 1,008 controls. Treatment of psoriasis using methotrexate or ustekinumab for any of the three discreet time periods was not associated with an increased risk of malignancy compared with no exposure to these drugs. However, TNF inhibitors, when used longer term (12 months or longer) were associated with an increased risk of cancer (odds ratio [OR]1.54, 95% confidence interval [CI], range 1.10– 2.15, p=0.01). This risk for TNF inhibitors disappeared when the analysis solely focused on patients receiving these agents as monotherapy. Also, the use of TNF inhibitors for shorter exposure periods (<12 months) was not associated with an increased risk of malignancy.6
Update on Psoriasis Comorbidities: Now That We Know What We Know, How Does It Change What We Do
Based on the presentation by Joel M. Gelfand MD, MSCE, FAAD, from University of Pennsylvania Perelman School of Medicine, Philadelphia Pennsylvania
Medical literature on the topic of psoriasis comorbidities has been rapidly expanding since around the turn of the century. There is a strong body of evidence that patients with psoriasis are at increased risk for developing psoriatic arthritis, cardiovascular disease (e.g., myocardial infarction, stroke), Crohn’s disease, diabetes, metabolic syndrome, mood disorders (e.g., anxiety, depression, suicide), and a much rarer condition, T-cell lymphoma.7–14 Genes and loci associated with psoriasis overlap with those associated with other conditions, such as diabetes and cardiovascular diseases (PSORS2/3/4, CDKAL1, ApoE4, TNFAIP32), with the potential contribution of environmental risk factors such as obesity and smoking.
The pathophysiology of psoriasis includes an increased presence of antigens, activation of T-cells and T-helper cell Types 1 and 17 cytokines (Th1 and Th17), and possibly higher levels of C-reactive proteins. These immunological conditions make patients vulnerable to other diseases associated with inflammatory processes.7–13 Chronic inflammation has been associated with numerous conditions, such as atherosclerosis and thrombosis.9 Epidermal proliferation associated with psoriasis plaques can increase uric acid and exacerbate oxidative stress.7 Patients under treatment for psoriasis might be exposed to varying risks and benefits associated with drug therapies.11 Finally, psoriasis in and of itself is associated with an adverse psychosocial impact owing to its disfiguring nature and chronic persistence.14 All of these factors combine such that psoriasis is comorbid with numerous conditions.
Cardiometabolic diseases. In a four-year, population-based cohort study from the United Kingdom (n=8,124 patients with psoriasis and 76,599 controls),15 patients with psoriasis had a hazard ratio for development of Type II diabetes mellitus of 1.64 (95% CI, 1.23– 2.18) if more than 10 percent of their BSA was affected. For each 10-percent increase in BSA affected by psoriasis, there was a 20-percent increase in risk for developing diabetes. This is not trivial: about 125,650 new cases of diabetes every year can be directly linked to psoriasis.15
Cardiovascular diseases. A prospective database study (n=130,976 patients with psoriasis and 556,995 control patients) of 5.4 years duration found patients with psoriasis had a higher rate of myocardial infarction than controls and exhibited a dose-response effect, in that those with the most severe psoriasis had the highest rate of myocardial infarction.7 A cohort study using the General Practice Research Database (N=3,603 patients with psoriasis) found that patients with severe psoriasis were at increased risk for cardiovascular death, independent of the traditional cardiovascular risk factors.16 Recent studies suggest that elevated risk for cardiovascular conditions might be attributed to arterial stiffness, an early independent predictor of adverse cardiovascular events. Arterial stiffness occurs frequently among psoriasis patients.17
Stroke. Stroke is a major cause of morbidity and mortality, and its associations with diabetes, hypertension, atrial fibrillation, transient ischemic attacks, and smoking are well known.18 In a population-based cohort study, it was found that severe psoriasis increased an individual’s chance of having a stroke by 44 percent. There is an elevated risk for stroke for all patients with psoriasis, but the risk for patients with mild psoriasis was deemed modest.9
Cancer. Patients with psoriasis are at an increased risk of cancer, particularly, but not exclusively, NMSC. It remains unclear whether new psoriasis treatments targeting interleukin (IL) 12 and 23 affect the patient’s risk for cancer.19 IL-12 and IL-23 play a role in the cellular responses and inflammatory processes associated with psoriasis but they are also involved in the immune response to tumors and infections. A systematic review and meta-analysis found that individuals with a history of cancer who developed psoriasis (or other immune-related disorders such as rheumatoid arthritis and inflammatory bowel disease) and were treated with TNF inhibition did not show an elevated risk for cancer recurrence or a new cancer.20 A review of cancer rates among patients with various immune-mediated diseases found that there was not an increased rate of cancer among patients with psoriasis in general or among patients with psoriasis who were on TNF inhibition therapy.21
Liver disease. Patients with psoriasis are at increased risk for all liver disease, nonalcoholic fatty liver disease (NAFLD), and cirrhosis. The prevalence of liver disease and cirrhosis was shown to increase in proportion to the BSA affected by psoriasis.22 The prevalence of NAFLD is estimated to be 20 to 60 percent among patients with plaque psoriasis.23–27 Nonalcoholic steatohepatitis (NASH) is a progressive form of fatty liver disease with an incidence of about 22 percent among patients with psoriasis.26 This area warrants further study.
Emerging comorbidities. Infections. A new area of research explores the risk of serious infection and opportunistic infection in patients with psoriasis.28 The risks of opportunistic infections and herpes zoster have been shown to be elevated among patients with moderate-to-severe psoriasis and could be statistically associated with immunosuppressive therapy.29
Sleep disorders. Psoriasis adversely affects sleep and has been associated with comorbid obstructive sleep apnea.30 It is thought that intermittent hypoxia caused by sleep apnea along with sleep deprivation creates a chronic inflammatory state that can set the stage for autoimmune disorders such as psoriasis.31
Chronic obstructive pulmonary disease (COPD). COPD shares some risk factors with psoriasis, including obesity, smoking, a sedentary lifestyle, and an association with metabolic syndrome, as well as potentially shared biological pathways such as Th17 driven inflammation.32 Patients with psoriasis have increased risk for impaired pulmonary function compared with control patients, as evidenced on spirometry.33 Patients with psoriasis also have a greater risk of developing COPD, compared with the general population, with an OR of 1.90 (95% CI, 1.36–2.65), and this risk increases with the severity of the psoriasis (2.15 for severe psoriasis, 95% CI, 1.26–3.67).34
Chronic kidney disease and end-stage renal disease (ESRD). Chronic kidney disease and ESRD have also been associated with psoriasis, according to a systematic review and meta-analysis (n=199,808), with an estimated pooled risk ratio for patients with psoriasis and chronic kidney disease of 1.34 (95% CI, 1.14–1.57) and psoriasis and ESRD of 1.29 (95% CI, 1.05–1.60).35 Severe psoriasis confers a greater risk than milder forms of psoriasis, with an adjusted hazard ratio of 1.90 (95% CI, 1.33–2.70) for chronic kidney disease and 2.97 (95% CI, 1.72–5.11) for ESRD.36
Peptic ulcer disease. Patients with psoriasis are at a greater risk than patients without psoriasis for developing peptic ulcer disease (adjusted OR 1.27, 95% CI, 1.03–1.58).37
Sexual dysfunction. Patients with psoriasis are more likely than the general population to experience sexual dysfunction, in particular erectile dysfunction (ED).38 According to one study,39 prevalence of ED was much higher in male patients with psoriasis (n=135, 61.5%) compared with controls (n=201, 48.3%), (p=0.001).
Drug therapies. TNF inhibitors. The systemic inflammation associated with psoriasis, psoriatic arthritis, and rheumatoid arthritis accelerates atherosclerosis and, in this way, might promote cardiovascular disorders. This risk exists even in patients with psoriasis who have no other cardiovascular risks, and cardiovascular disorders are especially prevalent in those with psoriasis and other cardiovascular risk factors. Thus, treatment of the underlying inflammation has been proposed to mitigate the cardiovascular risk.40 In a systematic review on rheumatoid arthritis,41 TNF inhibitors, such as infliximab, etanercept, and adalimumab, have been shown to be cardioprotective. In a meta-analysis of studies assessing the effect of TNF inhibitors on adverse cardiovascular events in patients with psoriasis, with or without psoriatic arthritis (n=49,795, 5 studies),42 the risk for a cardiovascular adverse event was significantly lower when patients were treated with TNF inhibitors compared with topical treatments or phototherapy (risk ratio 0.58, 95% CI, 0.43–0.77, p<0.001). In this meta-analysis,42 278 patients in the TNF inhibitor group and 1,063 in the control group had suffered myocardial infarctions. TNF inhibitors appeared to significantly reduce cardiovascular adverse events compared with methotrexate (risk ratio 0.67, 95% CI, 0.52–0.88, p=0.003). The risk for myocardial infarction specifically was significantly decreased, compared with topical therapy or phototherapy, among patients treated with TNF inhibition (risk ratio 0.73, 95% CI, 0.59–0.90, p=0.003) or with methotrexate (risk ratio 0.65, 95% CI, 0.48–0.89, p=0.007). The role of TNF inhibitors compared with other treatments for all-cause mortality and stroke remains to be elucidated. It must be noted that TNF inhibitors do not reduce the rate of heart failure and, in fact, might worsen it.43–45
Biologic therapies. In a systematic review and meta-analysis of 38 randomized clinical trials of adults with plaque psoriasis (n=18,024),46 the use of biologic therapies overall was not associated with any significant differences in the risk for major adverse cardiovascular events (MACEs), with an OR of 1.45 and 95-percent CI of 0.34–6.24. Specifically, for TNF alpha inhibitors (adalimumab, etanercept, infliximab), the OR was 0.67 (95% CI, 0.10–4.63); for anti-IL-17A agents (secukinumab and ixekizumab), the OR was 1.00 (95% CI, 0.09–11.09), and ustekinumab had an OR of 4.48 (95% CI,
0.24–84.77). There were no statistically significant differences among these various agents at approved doses compared with placebo.46
In light of what we know now, how should psoriasis be treated? People with psoriasis face a greater mortality burden compared with the general population.47 In a study from Noe and colleagues,48 BSA affected by psoriasis conferred a mortality risk such that patients with more than 10-percent affected BSA had a higher risk of all-cause death than those with less than three-percent BSA involvement.48 For many conditions, the more severe the psoriasis, the greater the comorbid risk to the patient. Thus, moderate and severe cases of psoriasis warrant special consideration.
Since patients with psoriasis are at increased risk for a number of cardiovascular conditions, dermatologists should educate these patients with psoriasis about the increased risk for cardiovascular disease and screen them according to standard guidelines for hypertension, diabetes, and hyperlipidemia. At present, fewer than half of all dermatologists ever conduct such screenings, but it is important since these patients might not be seeing other physicians or these other physicians might be unaware of a patient’s psoriasis and the risk it poses.49–51 Thirty-seven percent of newly screened patients with psoriasis were found to be at high risk for cardiovascular disease, and almost half of them (46%) were not optimally managed for these risk factors.49–51 The question that emerges from our growing knowledge of psoriasis comorbidities is whether more aggressive treatment of psoriasis would lower the risk of cardiovascular disease, cardiometabolic disorders, and overall mortality. Moreover, which type(s) of pharmacological therapy would be most helpful in reducing the risk of MACE and cardiovascular mortality? Early observational data suggest that methotrexate52,53 and TNF inhibitors54–56 are able to lower the risk of certain cardiovascular events (but not heart failure). Data are not yet available on whether phototherapy, topical treatments, and specific drugs such as apremilast, ustekinumab, secukinumab, ixekizumab, and guselkumab confer any cardioprotective benefits to patients with psoriasis. Randomized, controlled clinical studies and further systematic reviews and meta-analyses are needed to guide prescribers with more definitive evidence.
Treatment of atherosclerosis focuses mainly on reducing plasma lipid levels, but biomarkers for inflammation (high-sensitivity C-reactive protein and IL-6) have likewise been associated with an elevated risk for cardiovascular events independent of cholesterol level. Since statin drugs reduce both cholesterol levels and inflammation, it is unclear whether the reduction of vascular inflammation alone (without concomitant reduction of lipids) can reduce cardiovascular events associated with atherosclerosis. Canakinumab is a fully human monoclonal antibody that targets IL-1 beta and has known anti-inflammatory effects. The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS)57 was a randomized, double-blind, placebo-controlled clinical trial enrolling 10,061 patients with a prior myocardial infarction in order to determine if canakinumab could prevent recurrent vascular events in patients with a high-sensitivity C-reactive protein level of 2mg/L or greater. At 48 months, the median reduction from baseline in C-reactive protein levels was 26-percent, 37-percent, and 41-percent greater in patients who received 50, 150, or 300mg of canakinumab, respectively, compared with placebo.57 IL-6 levels also decreased, measured at 12 months. Canakinumab had no significant effect on either low-density lipoproteins (LDL) or high-density lipoproteins (HDL) cholesterol levels and resulted in a median of about five-percent elevation in triglycerides. The primary endpoint of the study was a composite endpoint of nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death; the composite endpoint occurred significantly less often among the 150mg canakinumab group compared with placebo (p=0.0275). Patients in the canakinumab group were more likely to experience neutropenia than those in the placebo group, and the pooled canakinumab group had significantly higher mortality attributable to infection or sepsis (incidence rate was 0.31 compared to 0.18 per 100 person-years, p=0.02).55 IL-1 beta inhibition appears to be an important anti-inflammatory pathway that might offer a degree of cardioprotection, but it is associated with its own risks for infection.57
Other studies are ongoing in evaluating the role of vascular inflammation and lipid metabolism in moderate-to-severe psoriasis. The Cardiovascular Inflammation Reduction Trial (CIRT)58 will evaluate methotrexate’s role in the possible reduction of major vascular events in a population without psoriasis but with a history of myocardial infarction, diabetes, or metabolic syndrome. TNF inhibition does not appear to reduce vascular inflammation compared with placebo at 16 weeks.59 The PSOLAR data do not show that biologics reduce the risk of MACE in patients with moderate-to-severe psoriasis.60
The association between psoriasis and cancer exists but remains to be more fully elucidated. As a precautionary measure, patients with psoriasis should be advised to stay current with all age-appropriate cancer screenings (e.g., colon, cervical breast, prostate, and lung). The incidence of smoking is higher among patients with psoriasis than the general population, so low-dose computed tomography (CT) screenings for lung cancer might be appropriate for patients 55 to 80 years of age with a history of smoking 30 or more packs of cigarettes per year as well as for current smokers and smokers who have quit in the last 15 years. Since psoriasis is associated with sexual dysfunction, clinicians might wish to ask patients about their sexual health.61 Lesions in the genital area can be particularly distressing and should be discussed with patients.62 Since over 45 percent of patients with genital-area psoriasis never discuss the problem with a clinician, the dermatologist might need to initiate such conversations.63 Patients with psoriasis are at increased risk for infections, so patients with guttate flares should be screened for possible streptococcal infection. Patients with severe psoriasis should also be screened for human immunodeficiency virus (HIV). Patients with psoriasis should be regularly vaccinated for influenza, pneumonia, hepatitis B, human papilloma virus (ages 9– 26 years), and shingles (>50 years of age).64
Update on the Pathogenesis of Psoriasis: A Bench-to-Bedside Success Story
Based on the presentation by Andrew Blauvelt, MD, MBA, Oregon Medical Research Center, Portland, Oregon
Psoriasis is caused by the complex interplay of four factors: autoantigens, environmental antigens, genetics, and the immune system. Autoinflammation, an inflammatory state not mediated by circulating autoantibodies and autoreactive T-cells, has recently been implicated as well.65 Genetics remains the key to better understanding the immune system and the pathogeneses of all autoimmune disorders, including psoriasis. Better understanding of the pathogenesis of psoriasis will allow for more accurate mapping of psoriasis pathways and improved ability to identify therapeutic targets, which, in turn, will lead to safer and more effective treatments.
The genetics of psoriasis. Psoriasis has strong heritability, and over 70 susceptibility loci/genes associated with psoriasis have been identified to date.66 In monozygotic twins, there is approximately a 70-percent genetic concordance, and 10 percent of the children of parents with psoriasis will develop psoriasis themselves (a 4- to 5-fold increase over general population). In the 1970s, psoriasis was found to be associated with the HLA Class I antigens, with associations between psoriasis and HLA-C (HLA-Cw6 in particular) and HLA-B alleles.67 The most common and dominant gene association is HLA-C*0602, but only 41 percent of people with psoriasis are HLA-C*0602-positive.68 In fact, the HLA-C*0602 gene, in and of itself, is neither necessary nor sufficient to develop psoriasis. About 20 percent of the population are carriers of the HLA-C*0602 gene, and about 10 percent of carriers develop psoriasis.66
Autoantigens. Antigens are molecules that induce an immune response in a host organism, and antibodies are the body’s method to target these antigens. Antigens are typically peptides, polysaccharides, and lipids that originate endogenously or exogenously. Autoantigens are endogenous antigens that are produced by normal cell metabolism or as a result of a viral or bacterial infection. The major histocompatibility (MHC) Class I antigen pathway serves to alert the immune system of the presence of a cell with a viral infection. MHC molecules are expressed on nucleated cells and platelets. In humans, HLA-A, HLA-B, and HLA-C correspond to MHC Class I, while HLA-DP, HLA-DM, HLA-DOA, HLA-DOB, HLA-DQ, and HLA-DR correspond to MHC Class II. Thus, autoantigens are endogenous proteins that can be attacked by the immune system, which is the reason they are associated with autoimmune disorders.
HLA-C*0602 is an MHC Class I molecule that normally presents autoantigens to CD8 T-cells. The autoantigens are ADAMTSL5 and LL-37. ADAMTSL5 is a peptide derived from the protein ADAMTSL5 and produced by melanocytes that can bind to HLA-C*0602 and stimulate psoriatic CD8+ T-cells. LL-37 is an antimicrobial peptide produced by keratinocytes, and it contains a VRSRRCLRL-like peptide that stimulates psoriatic T-cells.66
Environmental antigens also play a role in psoriasis. Streptococcal M proteins contain peptides that overlap with hyperproliferative keratin (K16/K17) peptides; these Streptococcal M proteins can stimulate psoriatic T-cells and have been implicated in certain cases of guttate psoriasis as environmental antigens. Yeast proteins are known to contain a VRSRRCLRL-like peptide that can stimulate psoriatic T-cells, and these exogenous antigens have been implicated in cases of beer-exacerbated psoriasis.66
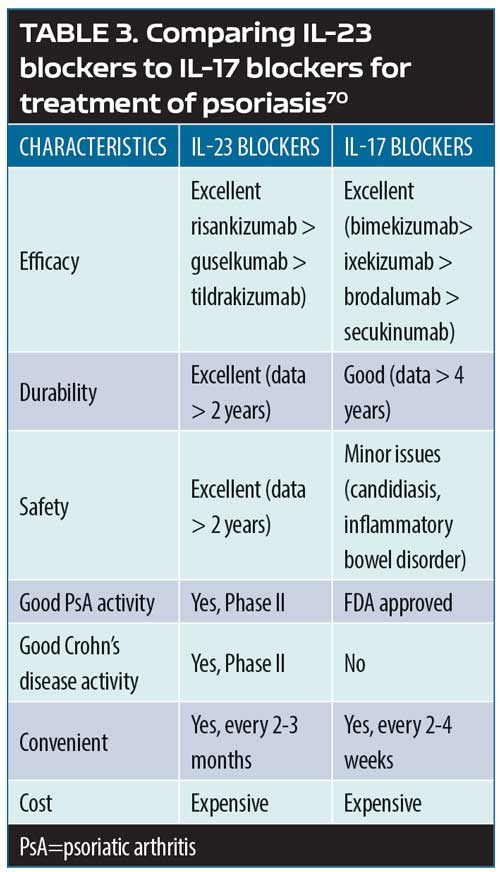
The psoriasis pathways: targets and mechanisms of actions. Psoriasis can be defined as a T-cell-mediated disease mainly driven by pathogenic T-cells that produce high levels IL-17 in response to IL-23. Greater understanding of the IL-23/T-17 axis, identified as the main pathway for psoriasis, has revolutionized the way psoriasis is treated today by offering important targets for pharmacotherapeutic interventions.69 Broadly blocking this pathway (e.g., with methotrexate) might work, but it might not be as safe or effective as a narrower blockade (e.g., TNF inhibition), which targets inflammation more specifically and results in greater efficacy and greater patient safety. However, if the target could be even more narrowly considered—that is, targeting psoriatic inflammation specifically along the IL-23, IL-17A, and IL17RA pathways—this should, in theory at least, allow for even greater safety and better effectiveness. Selective IL-23 blockers include guselkumab, tildrakizumab, and risankizumab. Selective IL-17A blockers include secukinumab, ixekizumab, and bimeizumab. Brodalumab blocks IL-17RA. Thus, pharmacological targets can be viewed as a continuum, with IL-23 the most upstream target and IL-22 and IL-17A downstream. While all of these points represent pharmacologically sound therapeutic targets, there are clinical differences among the inhibitors for these targets (Table 3).69,70
Not all of the cells that produce IL-17A are regulated by IL-23. Some cells, like Th17, depend on IL-23 in order to produce IL-17A, but others produce IL-17A independently of IL-23. This means that IL-23 inhibition will affect some IL-17A producers (those that depend on IL-23) but not the IL-23-independent IL-17A producers. Furthermore, it is thought that some of the IL-17A produced by cells other than Th17 in the skin and the gut might protect the body from candidiasis and inflammatory bowel disease. This would explain the association of candidiasis and inflammatory bowel disorder with IL-17 but not IL-23.69
As a general rule, the pharmacodynamic effects of a drug greatly exceed its pharmacokinetic effects, with the result being that a drug might have an impact on the underlying disease long after it has been eliminated from the body. For example, IL-23 inhibition might also cause the death of pathogenic skin-resident memory T17 cells, which depend on IL-23 for survival. Thus, the prolonged drug effect in destroying these T17 cells might lead to more durable psoriasis control.69
In a study evaluating clinically resolved psoriatic lesions,71 it was found that the lesions contained psoriasis-specific IL-17-producing alpha beta T-cell clones. Healed psoriatic skin (treated with phototherapy and etanercept) showed oligoclonal populations of T-cells, which produce IL-17. This observation might explain why new psoriatic lesions tend to recur at the same site as healed lesions.71
Update on Psoriatic Arthritis and the Focus on New Treatments
Based on the presentation by Arthur Kavanaugh, MD, Professor of Medicine and Director of the Center for Innovative Therapy (CIT) at University of California in San Diego, California
Psoriasis is the most prevalent auto-immune disease in the United States, with 2.2 percent of the population affected (7.5 million), of whom approximately 10 to 30 percent will develop psoriatic arthritis. Psoriatic arthritis can develop at any age but onset typically occurs between the ages of 30 and 50 years.72 The clinical presentation of psoriatic arthritis varies widely among patients, and, to this end, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) established six domains for treatment of psoriatic arthritis (not all of which affect each patient): peripheral arthritis, axial disease, enthesitis, dactylitis, skin disease, and nail disease.73 Since patients with psoriatic arthritis have different clinical presentations with different domains affected, an individualized therapeutic approach is required. Periodic re-assessment is necessary as psoriatic arthritis is a chronic, progressive condition. Further complicating the situation is that psoriatic arthritis, like plaque psoriasis, is associated with numerous comorbidities, including mental health disorders (depression, anxiety), obesity, Type II diabetes mellitus, osteoporosis, cardiovascular conditions, and cancer.74
The pharmacoeconomic implications of psoriatic arthritis. In a national cohort study from Denmark,75 10,525 patients with psoriatic arthritis and 20,777 matched controls were evaluated for employment, comorbidities, and costs to the healthcare system and society. Patients with psoriatic arthritis had significantly more comorbid conditions than controls, including cardiovascular disease, respiratory disease, and infectious disease, higher total healthcare costs, and lower incomes than controls. The relative risk (RR) of disability for patients with psoriatic arthritis progressed from an RR of 1.36 at five years prior to diagnosis (95% CI, 1.24–1.49) to an RR of 1.60 (95% CI, 1.49– 1.72) at the time of diagnosis to an RR of 2.69 10 years following diagnosis (95% CI, 2.40–3.02).75 A study in the United States comparing 5,492 matched pairs of patients with psoriatic arthritis and controls found the most common comorbidities were hyperlipidemia, hypertension, and diabetes and that patients with psoriatic arthritis had significantly more prescriptions, inpatient admissions, emergency department visits, and outpatient visits than controls and significantly higher total costs, pharmacy costs, and medical costs than controls.76 The total annual estimated healthcare burden of psoriasis overall could be over $35 billion (including direct costs plus lost productivity).77 The severity of psoriasis and psoriatic arthritis increases the cost.78
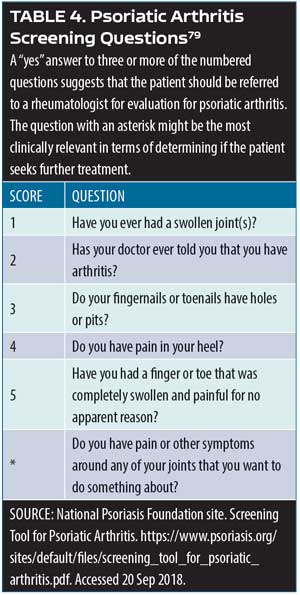
Screening psoriasis patients for psoriatic arthritis. Patients with psoriasis should be assessed periodically for joint pain, as it is a characteristic symptom of psoriatic arthritis. A simple assessment tool involves the use of a patient diagram (which can be an outline of human figure) where patients can mark the particular areas that cause them pain.79 Ibrahim and colleagues have defined five fundamental questions to be used in this assessment following identification of joint pain sites (Table 4).79 An important additional question to this screening instrument is: Do you have pain or other symptoms around any of your joints that you want to do something about? Clinicians might also wish to consider other psoriatic arthritis symptoms in this evaluation, such as joint swelling or tenderness, pain on movement, loss of function, and limitations to range of motion.
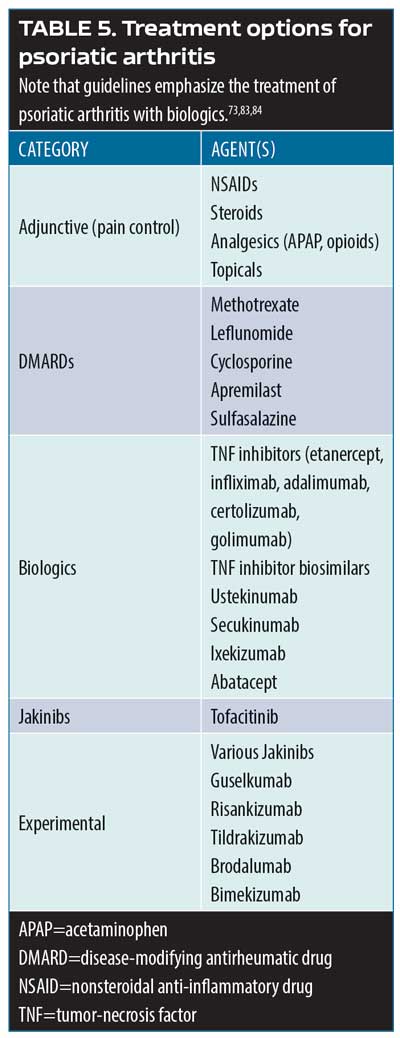
Psoriatic arthritis treatment options. Over 50 investigation monoclonal antibodies are being currently evaluated in late-stage clinical trials, and it is anticipated that at least half a dozen could be approved for market release in the coming year.80 The most important new developments in psoriatic arthritis involve these and other new treatment options. The main cytokine targets for the treatment of psoriatic arthritis are TNF, IL- 22/23, IL-17, and jakinibs (Table 5).81–84 Biologic agents serve to interrupt systemic inflammatory pathways.85
Golimumab. Golimumab is a human monoclonal antibody and TNF inhibitor that targets IL-6. In a Phase III, randomized, double-blind, placebo-controlled, multicenter clinical trial (GO-VIBRANT),83 480 patients were randomized to two groups: one received intravenous (IV) placebo (n=239) and the other IV golimumab at 2mg/kg (n=241) at Weeks 0, 4, 12, and 20. American College of Rheumatology 20-percent improvement criteria (ACR20) at 14 weeks was the primary endpoint and achieving at least PASI75 at 14 weeks was among the secondary endpoints. By Week 14, 75.1 percent of the patients in the golimumab group had achieved ACR20 versus 21.8 percent of the patients in the placebo group (p<0.001); at this time, 59.2 percent of the golimumab group compared with 13.6 percent of the placebo group had met the PASI75 endpoint (p<0.001). Adverse events were similar between groups, with 40.6 percent of placebo group and 46.3 percent of golimumab group having had at least one adverse event over the course of 24 weeks. The most commonly reported TEAE was infection (20.0% in the golimumab group compared with 13.8% in placebo group). No opportunistic infections or tuberculosis were reported in the study. Infusion site reactions were mild and rare (<2%). Discontinuation due to an adverse event was similar between groups, with 1.3 percent of the placebo group and 2.1 percent of the golimumab group leaving the study. Two patients died during the course of the study, both of whom were in the placebo group.86
The GO-VIBRANT study was preceded by the GO-REVEAL study,87 which found that patients with psoriatic arthritis exhibited greater improvement in signs and symptoms at 14 weeks with subcutaneous golimumab 50mg or 100mg versus placebo. ACR20 was achieved at 14 weeks by 48 percent of all patients in the golimumab group (51% of the 50mg and 45% of the 100mg group) versus nine percent of the patients in the placebo group (p<0.001). In this study, 74 percent of patients had at least three-percent BSA involvement with psoriasis at baseline; at 14 weeks, 40 percent of the golimumab 50mg group, 58 percent of golimumab 100mg group, and three percent of the placebo group achieved PASI75 (p<0.001). Golimumab was well-tolerated by patients.87
TNF-inhibition persistence. In five-year registry of patients with a rheumatologist’s diagnosis of psoriatic arthritis at the point they received their first TNF inhibitor, 46.7 percent of these patients were still taking the initially prescribed TNF inhibitor at five years. Factors associated with better five-year persistence were male sex, the use of etanercept or adalimumab rather than infliximab, and the lack of comorbid conditions at baseline. Estimates of five-year persistence for the first, second, and third TNF inhibitor were 53 percent (range 49–57%), 60 percent (range 43– 57%), and 48 percent (range 36– 59%), respectively.88 This suggests that there is good long-term persistence with TNF inhibition therapy.
Subcutaneous secukinumab. Secukinumab is a human monoclonal antibody that selectively targets IL-17A. In a randomized, double-blind, parallel-group clinical study (FUTURE 5),89 996 patients with psoriatic arthritis were randomized to receive either 300mg subcutaneous (SQ) secukinumab with a loading dose (n=222), SQ secukinumab 150mg with a loading dose (n=220), SQ secukinumab 150mg without a loading dose (n=222), or placebo (n=332) over 24 weeks. At 16 weeks, ACR20 response was achieved by 62.6 percent of patients in the secukinumab 300mg group, 55.5 percent of patients receiving secukinumab 150mg with loading dose, 59.5 percent of patients in the group who received secukinumab 150mg without loading dose, and 27.4 percent of patients in the placebo group (p<0.0001 for all doses compared to placebo). ACR50 and ACR70 response rates were significantly higher at Week 16 for all secukinumab groups compared to placebo. The rate of adverse events was similar across groups (any secukinumab group 56.3% vs. placebo 62.0%; the highest rate of adverse events occurred in the secukinumab 300mg group at 63.1%). The most frequently reported adverse events were upper respiratory tract infection, dyslipidemia, headache, hypertension, diarrhea, hypercholesterolemia, and urinary tract infection. However, most of these adverse events were single events. Results from FUTURE 5 suggest that secukinumab delivered with a loading dose and maintenance dose or at high doses of 300mg more significantly reduced joint damage associated with psoriatic arthritis than placebo or secukinumab without a loading dose. In fact, the 300mg dose of secukinumab provided numerically improved responses compared to the 150mg secukinumab dose with or without a loading dose.89
Apremilast. Apremilast is a phosphodiesterase-4 inhibitor that was shown to be effective in the treatment of psoriatic arthritis in the Psoriatic Arthritis Long-Term Assessment of Clinical Efficacy (PALACE) Phase III pivotal clinical trials (PALACE-1, PALACE-2, and PALACE-3).90 In all three PALACE studies, ACR20 was achieved at 16 weeks by significantly more patients taking apremilast 20mg or 30mg twice a day (BID) than placebo, with results durable out to 52 weeks. Apremilast was generally well tolerate by patients with the most commonly reported adverse events being diarrhea and nausea. In a meta-analysis of apremilast in patients with psoriatic arthritis,91 significantly more patients in the apremilast group achieved ACR20 at 16 weeks versus placebo. Data from the PALACE-1 trial has been collected for five years and grouped by patient response (ACR20, ACR50, and ACR70). At five years, 28.4 percent of patients achieved ACR70 and 46.9 percent achieved PASI75. Results are durable and trend toward increasing improvement.
Risankizumab. Risankizumab is a human monoclonal antibody that targets the p19 subunit of IL-23 and, in that way, indirectly inhibits the IL-23 pathway. In 2017, a Phase II study by Papp and colleagues92 reported that selective IL-23 blockade with risankizumab resulted in a superior clinical response in patients with moderate-to-severe plaque psoriasis (not psoriatic arthritis) compared with ustekinumab. In a Phase II study of 185 psoriatic arthritis patients,93 subcutaneous risankizumab 150mg and 75mg were compared to placebo. Patients were stratified by prior use of TNF inhibitors and concurrent methotrexate use. In this study, 49.4 percent of patients had skin psoriasis of three percent or greater BSA. At 16 weeks, significantly more risankizumab patients achieved ACR20 (57.1–65.0%) versus placebo (37.5%), and significantly more patients in the risankizumab group achieved PASI75, PASI90, and PASI100 than patients in the placebo group. TEAEs were similar among groups with the most frequently reported side effect being infection; no deaths and no cases of tuberculosis were reported. One MACE occurred in the risankizumab group.93
Tofacitinib. Tofacitinib is a Janus kinase inhibitor that was evaluated in a Phase III non-inferiority clinical study in 1,106 patients with moderate-to-severe plaque psoriasis.94 Patients were randomized to receive either tofacitinib (5mg or 10mg BID) or etanercept (50mg twice weekly). Tofacitinib 10mg BID was non-inferior to etanercept 50mg twice weekly; the same did not apply to tofacitinib 5mg BID. In this 12-week study, 58.8 percent of the etanercept group achieved PASI75 compared to 63.6 percent of the tofacitinib 10mg BID group, and 39.5 percent of the tofacitinib 5mg BID groups achieved PASI75 at 12 weeks versus 5.6 percent of the placebo group.94
A 12-month, double-blind, active- and placebo-controlled Phase III trial compared tofacitinib, adalimumab, and placebo among 317 patients with psoriatic arthritis who had an inadequate clinical response to DMARD therapy.95 Patients were randomized to oral tofacitinib 5mg BID, oral tofacitinib 10mg BID, SQ adalimumab 40mg every two weeks, or placebo with a blinded switch at three months to tofacitinib 5mg or 10mg. ACR20 response rates at three months were 50 percent and 61 percent for the tofacitinib 5mg and 10mg groups, respectively, compared to 33 percent for the placebo group and 52 percent for the adalimumab group. Adverse event rates for 12 months were 66 percent and 71 percent in the tofacitinib 5mg and 10mg groups, respectively, 72 percent in the adalimumab group, and 69 percent and 64 percent in the placebo groups switched at three months to tofacitinib 5mg and 10 mg, respectively. Thus, tofacitinib was shown to be significantly superior to placebo at three months in patients with psoriatic arthritis who had failed treatment with a disease-modifying antirheumatic drug (DMARD) treatment.
Psoriatic arthritis comorbidities. Psoriatic arthritis, like plaque psoriasis, is associated with numerous, potentially serious comorbid conditions that can lead to decreased function, complicate treatment, and reduce the patient’s quality of life. Among these comorbidities are obesity, infections, metabolic syndrome, Type II diabetes, inflammatory bowel disease, autoimmune ophthalmic disease, osteoporosis, cancer, fatty liver disease, kidney disease, cardiovascular disorders, and cancer.96 A comparison of patients with psoriatic arthritis (n=90) and 240 controls was made using coronary computed tomography angiography (CCTA) to evaluate atherosclerosis. Patients had no prior history or known cardiovascular disease; they underwent CCTA for chest pain or because they had risk factors for heart disease. Patients with psoriatic arthritis had an increased prevalence, greater burden, and more severe coronary atherosclerosis on CCTA than control patients. The prevalence of overall plaque in the coronary artery was 60 percent versus 35 percent (p<0.001) for psoriatic arthritis versus control groups. For calcified plaque, it was 32 percent versus 17 percent (p=0.002) for patients with psoriatic arthritis versus control patients; for mixed plaque 22 percent versus eight percent (p<0.001), for non-calcified plaque 43 percent versus 22 percent (p<0.001), and for combined mixed and non-calcified plaque 51 percent versus 26 percent (p<0.01). Three-vessel disease occurred significantly more frequently among patients with psoriatic arthritis than controls (13% vs. 3%, p<0.001) as did obstructive plaques (defined as >50% stenosis), which occurred in nine percent of patients with psoriatic arthritis versus three percent of control patients (p=0.033).97
New Developments in the Treatment of Moderate-to-severe Psoriasis: Biologics
Based on the presentation by Craig L. Leonardi, MD, Associate Clinical Professor of Dermatology, St. Louis University Medical School, St. Louis, Missouri
Cytokines, those small proteins that regulate specific activities of a wide range of immunocompetent cells, are only beginning to be elucidated. Their main known function can be described, perhaps simplistically, as regulating the communication among cells within and outside of the immune system. It is quite possible that not all cytokines have been discovered, but recent advances have identified several and their in-vivo functions. The main groups of cytokines are the pro-inflammatory/anti-inflammatory cytokines (described here), cytokines related to neutrophil and eosinophil recruitment and activation, Th cell cytokines, T-regulatory (Treg) cell cytokines, and cytokines involved in the recruitment and growth of T-cells. Signaling pathways within the nucleus and cytoplasm of the cell connect cytokine receptors; these pathways can activate cytokines (along with other factors) during or after transcription.98,99
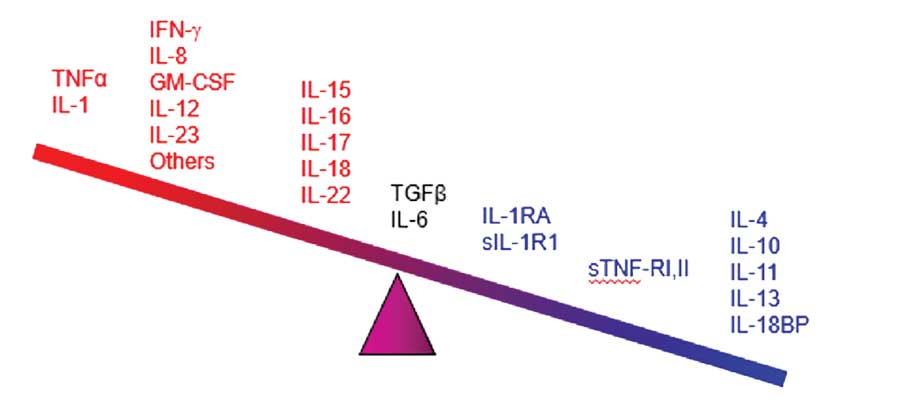
Cytokines appear to be the main mediators of the cutaneous inflammation associated with psoriasis.100 The cytokine network is extremely complex: cytokines can change the expression of receptors which, in turn, can change how responsive a source and/or target cell are to the cytokine. In treating psoriasis, targeting specific cytokines for inhibition has reduced chronic inflammation and led to major advances in controlling the disease. The main families of cytokines associated with psoriasis are TNF alpha, IL, and TGF beta.101 Humanized monoclonal antibodies (the -mab suffix drugs) target specific cytokines, and some have shown promise in the treatment of psoriasis. The cytokines associated with psoriasis appear in Figure 1, many of which have become important new targets for drug development.102
Certolizumab-pegol. Certolizumab-pegol (CZP) is a monoclonal antibody that targets TNF alpha. It is a PEGylated antigen-binding fragment (Fab) of a humanized TNF-inhibitor monoclonal antibody. CZP has been approved for use by the FDA for the indications of Crohn’s diseases (2008), rheumatoid arthritis (2009), active psoriatic arthritis (2013), ankylosing spondylitis (2013), and most recently for the treatment of psoriasis.103 The CIMPASI-1 (n=234) and CIMPASI-2 (n=227) placebo-controlled trials104 evaluated SQ CZP in patients with moderate-to-severe plaque psoriasis, scored based on the PASI and PGA scores, and found significant improvements in both active groups versus placebo at 16 weeks. In CIMPASI-2, patients were randomized to one of three treatment arms: 400mg SQ CZP every two weeks (n=87), 400mg SQ CZP at Weeks 0, 2, and 4 followed by 200mg every two weeks thereafter for 16 weeks (n=91), and placebo every two weeks (n=49). The primary endpoint of the study was meeting PASI75 at 16 weeks, which was achieved by 82.6 percent of the first group (400mg every 2 weeks), 81.4 percent of the second group (400/200mg), and 11.6 percent of placebo patients. The PGA endpoint called for improvement of at least two points by 16 weeks; 71.6 percent, 66.8 percent, and 2.0 percent met this endpoint in the first, second, and placebo groups, respectively. Thus, CZP demonstrated significant improvements from baseline at 16 weeks compared with placebo for both PASI75 and PGA endpoints. Adverse events were similar to those reported when using CZP for other approved indications.104,105
Guselkumab. Guselkumab is an IL23-P19 inhibitor and was evaluated in a 48-week, Phase III, double-blind, placebo-controlled VOYAGE 1 trial.106 Patients were randomized into one of three groups: 1) patients (n=329) received guselkumab 100mg (Weeks 0 and 4 and then every 8 weeks), 2) patients (n=174) started with placebo at Weeks 0, 4, and 12 and then rotated to guselkumab at Weeks 16 and 20 and every eight weeks thereafter, and 3) patients (n=334) received adalimumab 80mg at Week 0, 40mg at Week 1, then 40mg every two weeks through Week 47. Endpoints were the PASI score, the patient-reported outcomes on the Dermatology Life Quality Index, and the Psoriasis Symptoms and Signs Diary. Guselkumab demonstrated significant superiority (p<0.001) to placebo at 16 weeks (85.1% vs. 6.9% on Investigator Global Assessment Score [IGA]) and 73.3 percent versus 2.9 percent showed a 90-percent or greater improvement in PASI score over baseline. When compared to adalimumab, guselkumab was significantly superior (p<0.001) in IGA and PASI90 at Weeks 16, 24, and 48.106 During the placebo-controlled phase of the study, at least one adverse event was reported by 49.4 percent, 51.7 percent, and 51.1 percent of the placebo, guselkumab, and adalimumab groups, respectively; over the entire course of the study, 73.9 percent of the guselkumab group and 74.5 percent of adalimumab group reported at least one adverse event. In the group that transitioned from placebo to guselkumab, 64.8 percent of patients reported at least one adverse event during Weeks 16 to 48. Serious adverse events were reported by less than five percent of all patients; 4.9 percent of guselkumab and 4.5 percent of adalimumab patients reported at least one serious adverse event over the course of the study.106 In a Phase II study of guselkumab, ACR 20/50/70 scores showed the significant superiority of guselkumab over placebo. 107
Tildrakizumab. Like guselkumab, tildrakizumab targets IL-23 p19 and was shown in two Phase III clinical trials to be more effective than placebo and etanercept for treating moderate-to-severe plaque psoriasis.108 In these international multicenter, three-part, parallel-group, randomized, double-blind studies, patients were randomized to tildrakizumab 200mg, tildrakizumab 100mg, or placebo (reSURFACE-1 N=1,772) or to tildrakizumab 200mg, tildrakizumab 100mg, placebo, or etanercept 50mg (reSURFACE-2 , N=1,090). In the reSURFACE-1 study, at Week 12, 192 patients (62%) in the 200mg group and 197 patients (64%) in the 100mg group achieved PASI75, compared with nine patients (6%) in the placebo group. In the reSURFACE-2 study, at Week 12, 206 patients (66%) in the 200mg group, and 188 patients (61%) in the 100mg group achieved PASI75, compared with nine patients (6%) in the placebo group and 151 patients (48%) in the etanercept group. Serious adverse events were similar and low in all groups in both of these trials. One patient died in reSURFACE-2 (tildrakizumab 100mg group) but the cause of death could not be determined. The FDA approved tildrakizumab for market release in March 2018 for the treatment of plaque psoriasis.
Risankizumab. Risankizumab targets IL-23 p19, and the IMMhance trial evaluated its use against placebo over 16 weeks, but results are not yet published. From data on file at AbbVie, preliminary results found that 73.2 percent of risankizumab patients achieved PASI90 and 47.2 percent PASI100 at 16 weeks compared with 2.0 percent and 1.0 percent for placebo, respectively. In a recent study of 166 patients with moderate-to-severe plaque psoriasis,92 patients were randomized to receive subcutaneous risankizumab (one 18mg dose at Week 0 or 90 or 180mg doses at Weeks 0, 4, and 16, or ustekinumab (45mg or 90mg based on body weight at Weeks 0, 4, and 16). At Week 12, of the patients who had a 90-percent or greater reduction in their PASI score, 77 percent were in the risankizumab group (pooled 90mg and 180mg group results) versus 40 percent in the ustekinumab group (p<0.001). Results demonstrated durability up to 20 weeks for risankizumab. A serious adverse event was reported by 12 percent of patients in the risankizumab 18mg group, 15 percent in the risankizumab 90mg group, and eight percent ustekinumab, including one MACE and two BCCs. No serious adverse events were reported in the risankizumab 180mg group.92
Mirikizumab. A relatively new monoclonal antibody to be evaluated for the treatment of moderate-to-severe plaque psoriasis is mirikizumab, which targets IL-23 p19. A Phase II randomized, multicenter, double-blind, placebo-controlled trial is being conducted using different doses of mirikizumab and evaluating them for safety and efficacy is ongoing [ClinicalTrials.gov Identifier: NCT02899988]. Results of this study were presented at the 2018 American Academy of Dermatology Annual Meeting [Abstract # 6131] by Rich et al. In this study, 205 patients were randomized to mirikizumab 300mg every eight weeks (Q8W), 100mg Q8W, 30mg Q8W, or placebo Q8W over the course of 16 weeks. The mirikizumab 300mg and 100mg groups were significantly more likely to achieve PASI90 at 16 weeks compared to placebo (67% and 59%, respectively vs. 0 in placebo, p<0.001). Twenty-nine percent of the mirikizumab 30mg group achieved PASI90 at 16 weeks, but this was not significant compared to placebo. PASI100 scores were achieved by significantly more patients in the mirikizumab 300mg and 100mg groups than the placebo group at 16 weeks (31%, p=0.007 for both); 16 percent of the patients in the mirikizumab 30mg group achieved PASI100 at 16 weeks, which was significant versus placebo (0%, p=0.039). Pooling safety results from all mirikizumab groups, at least one TEAE was reported by 48.4 percent of mirikizumab patients compared with 48.1 percent of the placebo patients, and 1.3 percent of the mirikizumab patients and 1.9 percent of the placebo patients reported at least one serious adverse event.
Secukinumab. The IL-17 family of cytokines includes IL-17A, IL-17A/F, IL-17F, IL-17E, and IL-17C, and secukinumab targets IL-17A. There are long-term data for secukinumab showing that five-year PASI score improvements are durable over time.109 At Year 1, 168 patients entered the extension study, and at the end of Year 5, 126 patients completed 300mg (every 4 weeks) treatment. PASI 75/90/100 responses at Year 1 (88.9%, 68.5% and 43.8%, respectively) were sustained to Year 5 (88.5%, 66.4% and 41%, respectively). Results from Future 5, a placebo-controlled study, evaluated 300mg and 150mg of secukinumab and 150mg secukinumab with no loading dose compared with placebo,89 and in this study, significantly more patients achieved PASI75 and PASI90 with secukinumab compared to placebo.
Ixekizumab. Ixekizumab targets IL-17A, and the UNCOVER-3 trial provides long-term data with durable efficacy up through 108 weeks. In this randomized, controlled, Phase III study, 1,346 patients with moderate-to-severe plaque psoriasis were randomized to one of four treatment arms.110 Patients received ixekizumab 80mg every two or four weeks, etanercept 50mg twice a week, or placebo. Patients entered the long-term extension phase at Week 12 when all patients were switched to ixekizumab every four weeks.
The results of a head-to-head comparison of ixekizumab compared to ustekinumab111 reported that 52.2 percent more patients in the ixekizumab group achieved PASI100 scores at 52 weeks compared to those in the ustekinumab group (35.5%).
Brodalumab. In contrast to ixekizumab, which binds to IL-17, brodalumab binds to the IL-17 receptor and, in that way, prevents IL-17 from activating that receptor. In two Phase II studies comparing brodalumab to ustekinumab in patients with moderate-to-severe plaque psoriasis, brodalumab was associated with significant clinical improvements compared to ustekinumab.112 At 12 weeks, brodalumab 210mg resulted in 44 percent of patients achieving PASI100 scores compared with 22 percent of the patients taking ustekinumab in AMAGINE-2; these results were similar to AMAGINE-3, where 37 percent of patients in the brodalumab group achieved PASI100 scores compared to 19 percent of patients in the ustekinumab group (p<0.001, both). Rates of neutropenia were higher with brodalumab and ustekinumab versus placebo. Safety issues with brodalumab surfaced around this time when a numeric imbalance was noted in terms of cases of depression, suicidal ideation, and suicidal behavior. During the Phase III brodalumab trials, the manufacturer introduced psychological surveys to help detect patients at risk but the intervention was not successful. All trials were halted in the second quarter of 2015, but brodalumab was eventually approved by the FDA in 2017 with a black box warning.113 It should be noted in this context that, overall, patients with psoriasis appear to have higher rates of suicidal ideation than the general population. A 1993 survey found that 5.5 percent of patients with psoriasis had active suicidal ideation at the time of the study, and the more severe the patient considered his or her psoriasis, the more likely the patient was to have suicidal thoughts.114
Bimekizumab. Bimekizumab has a novel dual mechanism of action in that it neutralizes both IL-17A and IL-17F, two key pro-inflammatory cytokines.115 Based on clinical data on file with UCB, in the Phase II BE ABLE clinical study, 79 percent and 60 percent of bimekizumab patients achieved PASI90 and PASI100 scores, respectively, in 12 weeks. The safety and efficacy of bimekizumab was demonstrated in a first-in-human randomized clinical trial assessing patients with mild plaque psoriasis.116 Based on early results, this new agent appears to hold promise for the treatment of psoriasis.
The paradigm shift and future directions. The classic treatment of psoriasis had always been a stepwise progression from over-the-counter products to prescription topicals to phototherapy and then, if these treatments were no longer effective, finally to systemic therapy. In all cases, patients were expected to receive treatment in an orderly sequence, step by step, and not skip any steps. Aggressive therapy was reserved only for those patients who failed more conservative approaches. Today, the evolution of new drugs for psoriasis and the increased understanding of its pathogenesis have resulted in our discarding the classic paradigm in favor of a new approach. If a patient with psoriasis is an appropriate candidate for topical treatment, that should be the first-line approach; if that fails or the patient is not a good candidate for topical therapy, then there are three main types of therapy: biologic agents, traditional systemic agents (such as methotrexate), and phototherapy. The choice of therapy must be based on the individual characteristics of the patient. Within each group there are further therapeutic choices, again based on the needs of the individual patient. There is no longer any need for a patient to have to progress through and fail multiple treatment steps before receiving biologic therapy.
What’s New in Topical Therapy?
Based on the presentation by Linda Stein Gold, MD, Henry Ford Health System, Detroit, Michigan
Patients often struggle with identifying what topical products they are currently using or have used in the past (“that stuff in the tube with the red label” or “the white cream”) when they present to their dermatologist’s office. Likewise, the misconceptions clinicians have about topical products can interfere with the optimal use of these treatments. Topical treatments remain the mainstay of psoriasis treatment, and the introduction of new molecules and designer vehicles holds great promise for patients who benefit from topical psoriasis therapy. Among these clinical misconceptions are mistaken ideas about the vehicle. All topical therapy involves the union of an active drug(s) and a vehicle—both of which contribute to the product’s efficacy. A change in vehicle can result in differences in effectiveness and safety, as well as in patient acceptance and adherence. For example, calcipotriene is marketed as four products, each using a different vehicle: ointment, cream, foam, and scalp solution. Yet, the efficacy and level of skin irritation between the four products varies. Data from the package inserts indicates efficacy ranging from 31 percent (scalp solution) to 70 percent (ointment) of the subjects reporting marked improvement, and skin irritation occurring in as few as two percent (foam) and as many as 23 percent (scalp solution) of the subjects, following twice weekly applications over eight weeks.
Another potential clinical misconception involves the potency ratings of various topical products. Potency categorizations can also be misleading in that the potency does not necessarily predict efficacy of the product. For example, betamethasone dipropionate 0.05% has a variety of potency categorizations depending on the vehicle. Diprolene cream 0.05% and diprolene ointment 0.05% are both ranked as Class I potency, while diprosone lotion 0.05% is ranked as a Class V. Diprolene cream AF 0.05% and diprosone ointment 0.05% are both ranked as Class II potency, while diprosone cream 0.05% and betamethasone dipropionate emollient spray 0.05% are ranked as Class III. Desoximetasone 0.25% spray is Class I while desoximetasone 0.25% cream and ointment are both Class II.117
Clinicians prescribing topical treatments for their patients with psoriasis can rely on a robust armamentarium of products which, in turn, opens up the challenge of finding the right product for each individual patient. Patient education is also important so that patients understand how to use their treatments and how their treatments work, thus increasing their likelihood to adhere to their treatment plans.
“Designer vehicles” and topical treatments. The vehicle is the method by which the active ingredient is delivered into the skin. Recent advances allow for the precise measurement of drug concentrations to be distributed between skin layers and has led to coining a new word: dermatokinetics. Despite the promise of these advances, establishing the pharmacokinetics of topical drugs in the skin is still challenging. Skin biopsies allow for good assessments but are invasive, expensive, and impractical if used only to obtain kinetic data. An increasingly important in-vivo alternative technique is the vasoconstrictor assay (VCA) test.118 The VCA measures blanching effects of topical products as a surrogate marker for bioavailability. Skin blanching occurs as a result of vasoconstriction in the lower skin layers, and VCA measures if the drug has passed through the skin, but not necessarily whether the drug is within the skin. The “depot effect” of designer vehicles allows a steroid to penetrate the stratum corneum layer of skin and remain in position in the epidermis and dermis to topically treat psoriasis.
A good example of a designer vehicle and its role in optimal topical care is the novel medium-potency formulation of betamethasone dipropionate 0.05% emollient spray. This spray has shown similar efficacy when compared head-to-head with augmented (super-potent) betamethasone dipropionate 0.05% lotion in the treatment of moderate plaque psoriasis.119 This study randomized 351 patients with psoriasis to one of three groups: spray (DFD-01), lotion (diprolene), or vehicle alone. Successful treatment was defined a score of 0 or 1 on the IGA and an improvement of two or more grades from baseline. At eight days, 10 percent of the DFD-01 group, 6.7 of the diprolene group, and 1.2 percent of vehicle group achieved success (p=0.003 DFD-01 vs. vehicle). DFD-01 relieved signs of scaling and erythema earlier than diprolene or vehicle at Day 4 (p is less than or equal to0.048). All products were well tolerated but there was significantly more burning and stinging on application with diprolene compared with DFD-01 (13.6% vs. 4.1%, p=0.006).119
There is growing understanding regarding the role vehicles play in topical treatments, and the production of certain novel designer vehicles has led to the drug development question as to whether we can further improve the vehicle and create something that will allow even better penetration of the active agent into the skin. A prerequisite to improving a product’s bioavailability is good penetration of the stratum corneum. This, in turn, means that the active ingredient must be dissolved in the vehicle in order to assure good penetration of the stratum corneum. Adolf Fick’s work on diffusion set forth in 1855 still applies. Fick’s First Law describes the diffusive flux to the concentration under the assumption of the steady state and holds that the flux will go from regions of highest concentrations to regions of lowest concentrations with a magnitude proportional to the concentration gradient. Applied to topical therapeutic products, this means that the rate of skin penetration will be proportional to the amount of active agent dissolved in the vehicle. Vehicles with crystalline agents will not work as well because the agents are not dissolved into the vehicle. The transition to foam products with propellants (dimethyl ether and butane) has minimized the use of crystalline agents and enhanced the penetration with minimal systemic absorption. A study comparing the efficacy and safety of an aerosol foam of a fixed-combination of calcipotriene 0.005% and betamethasone dipropionate 0.064% in patients with psoriasis (n=376).120 found that significantly more patients using the aerosol product achieved the primary endpoint (“clear” or “almost clear” rated skin with at least a 2-step improvement over baseline) at four weeks compared to patients treated with the calcipotriene/betamethasone dipropionate ointment (54.6% vs. 43.0%, p=0.025). Tolerability was good and similar between groups
Tazarotene. Tazarotene, which is part of a non-isomerizable class of retinoic acid receptor (RAR)-specific retinoids, is indicated for use as a topical treatment for stable plaque psoriasis and is often used in combination with other therapies.121,122 Tazarotene reduces the proliferation of keratinocytes and has an anti-inflammatory effect. As a topical retinoid drug, it is classified as Pregnancy Category X and must be used with caution. About 10 to 30 percent of patients who use tazarotene experience side effects such as pruritus, burning or stinging sensations at application site, erythema, exacerbation of psoriasis, skin irritation, and skin pain. Combining tazarotene with corticosteroids can enhance efficacy while reducing the adverse effects.122
A fixed combination of tazarotene and halobetasol propionate in a lotion formulation has completed clinical trials.123 In two randomized, multicenter, double-blind, vehicle-controlled Phase III clinical trials (n=418), patients with moderate-to-severe plaque psoriasis were randomized to receive halobetasol propionate 0.01% and tazarotene 0.045% lotion (HP/TAZ) once a day over eight weeks versus vehicle alone. The primary endpoint was an IGA score of “clear” or “almost clear” and at least a two-grade improvement over baseline. By the second week, HP/TAZ lotion was significantly superior over the vehicle, and at eight weeks, 35.8 percent (Study 1) and 45.3 percent (Study 2) of the HP/TAZ lotion group met the primary endpoint, compared with 7.0 percent and 12.5 percent of vehicle alone group (p<0.001 for both). HP/TAZ was significantly more effective in reducing signs and symptoms of psoriasis compared to the vehicle alone. The most frequently reported treatment-related adverse events were contact dermatitis (6.3%), application site pain (2.6%), and pruritus (2.2%). Thus, HP/TAZ lotion appears to be an effective treatment option for controlling moderate-to-severe plaque psoriasis.123
New molecules on the horizon? Several new molecules are currently under investigation as potential agents in the topical treatment of psoriasis. Tapinarof (GSK2894512) acts as an agonist at the anyl hydrocarbon receptor (AhR), which might result in better regulation of the cytokine household and improved barrier function. As an AhR agonist, tapinarof is in a unique new class of molecules. A small open-label study of patients with atopic dermatitis found it to be effective;124 a 1% tapinarof cream was associated with fewer side effects than a 2% cream, but both had similar effectiveness. Tapinarof has also been shown to be effective in treatment of psoriasis.125 Currently, a dose-ranging study of tapinarof for the treatment of plaque psoriasis has been completed and promising preliminary results were presented during the American Academy of Dermatology scientific sessions.126
Many pro-inflammatory cytokines have been implicated in the pathogenesis of psoriasis via the Janus kinase (JAK) pathways. Thus, a new target for a topical product has been the down-modulation of certain key inflammatory cell markers by way of a JAK1/JAK2 inhibitor. A novel JAK1/JAK2 inhibitor has been developed that blocks the signal transduction of multiple pro-inflammatory cytokines. This new drug, INCB018424, is intended for the treatment of plaque psoriasis. Topical INCB018424 phosphate 1.0% or 1.5% cream was applied for four weeks in five sequential cohorts of five patients each and evaluated at 28 days, and was shown to be effective in modulating pro-inflammatory cytokines. 127
Conclusion
The field of dermatology allows clinicians to make a powerful positive impact on the lives of their patients, and these new medical breakthroughs, innovative treatments, new insights, and scientific findings are helping dermatologists provide better care for their patients. Maui Derm focuses on the changes in dermatology from year over year, allowing clinicians to better process this new knowledge and use it to change clinical practice for optimal dermatological outcomes.
References
1. Mariette X, Förger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77(2): 228–233.
2. 1Ryan C, Sadlier M, De Vol E, et al. Genital psoriasis is associated with significant impairment in quality of life and sexual functioning. J American Acad Dermatol. 2015;72(6):978–983.
3. Ryan C, Menter A, Guenther L, et al. Efficacy and safety of ixekizumab in a randomized, double-blinded, placebo-controlled Phase 3b study of patients with moderate-to-severe genital psoriasis. Br J Dermatol. 2018 May 10. doi: 10.1111/bjd.16736. [Epub ahead of print].
4. Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371(4):326–338.
5. Fiorentino D, Ho V, Lebwohl M, et al. Risk of malignancy associated with biologic therapy and methotrexate (MTX) in the Psoriasis Longitudinal Assessment and Registry (PSOLAR). Poster Abstract B221. Presented at the American Academy of Dermatology. Orlando, Florida. 3–7 March 2017. J Am Acad Dermatol. 2017;76:(6 Suppl).
6. Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77(5):845–854.e845.
7. Gelfand JM, Neimann AL, Shin DB, et al. Risk of myocardial infarction in patients with psoriasis. J Am Med Assoc. 2006;296(14):1735–1741.
8. Gelfand JM, Azfar RS, Mehta NN. Psoriasis and cardiovascular risk: strength in numbers. J Invest Dermatol. 2010;130(4):919–922.
9. Gelfand JM, Dommasch ED, Shin DB, et al. The risk of stroke in patients with psoriasis. J Invest Dermatol. 2009;129(10):2411–2418.
10. Gelfand JM, Yeung H. Metabolic syndrome in patients with psoriatic disease. J Rheumatol. 2012;89:24–28.
11. Lee MS, Lin RY, Lai MS. Increased risk of diabetes mellitus in relation to the severity of psoriasis, concomitant medication, and comorbidity: a nationwide population-based cohort study. Journal of the American Academy of Dermatology. 2014;70(4):691-698.
12. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: Implications for management. Journal of the American Academy of Dermatology. 2017;76(3):393-403.
13. Takeshita J, Grewal S, Langan SM, et al. Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol. 2017;76(3):377–390.
14. Connor CJ, Liu V, Fiedorowicz JG. Exploring the physiological link between psoriasis and mood disorders. Dermatol Res Prac. 2015;2015:409637.
15. Wan MT, Shin DB, Hubbard RA, et al. Psoriasis and the risk of diabetes: a prospective population-based cohort study. J Am Acad Dermatol. 2018;78(2):315–322.e311.
16. Mehta NN, Azfar RS, Shin DB, et al. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J. 2010;31(8):1000–1006.
17. Dregan A. Arterial stiffness association with chronic inflammatory disorders in the UK Biobank study. Heart (British Cardiac Society). 2018;104(15):1257–1262.
18. Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371(9624):1612–1623.
19. Ergen EN, Yusuf N. Inhibition of interleukin 12 and/or interleukin 23 for treatment of psoriasis: What is the evidence for an effect on malignancy? Exp Dermatol. 2018;27(7):737–747.
20. Micic D, Komaki Y, Alavanja A, et al. Risk of cancer recurrence among individuals exposed to antitumor necrosis factor therapy: a systematic review and meta-analysis of observational studies. J Clin Gastroenterol. 2017 Jul 21. doi: 10.1097/MCG.0000000000000865. [Epub ahead of print].
21. Chen Y, Friedman M, Liu G, et al. Do tumor necrosis factor inhibitors increase cancer risk in patients with chronic immune-mediated inflammatory disorders? Cytokine. 2018;101:
78–88.
22. Ogdie A, Grewal SK, Noe MH, et al. Risk of incident liver disease in patients with psoriasis, psoriatic arthritis, and rheumatoid arthritis: a population-based study. J Invest Dermatol. 2018;138(4):760–767.
23. Gisondi P, Targher G, Zoppini G, Girolomoni G. Non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol. 2009;51(4):758–764.
24. Miele L, Vallone S, Cefalo C, et al. Prevalence, characteristics and severity of non-alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol. 2009;51(4):778–786.
25. Madanagobalane S, Anandan S. The increased prevalence of non-alcoholic fatty liver disease in psoriatic patients: a study from South India. Australas J Dermatol. 2012 Aug;53(3):190–197.
26. Roberts KK, Cochet AE, Lamb PB, et al. The prevalence of NAFLD and NASH among patients with psoriasis in a tertiary care dermatology and rheumatology clinic. Aliment Pharmacol Ther. 2015 Feb;41(3):293–300.
27. van der Voort EA, Koehler EM, Dowlatshahi EA, et al. Psoriasis is independently associated with nonalcoholic fatty liver disease in patients 55 years old or older: results from a population-based study. J Am Acad Dermatol. 2014;70(3):517–524.
28. Wakkee M, de Vries E, van den Haak P, Nijsten T. Increased risk of infectious disease requiring hospitalization among patients with psoriasis: a population-based cohort. J Am Acad Dermatol. 2011;65(6):1135–1144.
29. Takeshita J, Shin DB, Ogdie A, Gelfand JM. Risk of serious infection, opportunistic infection, and herpes zoster among patients with psoriasis in the United Kingdom. J Invest Dermatol. 2018;138(8):1726–1735.
30. Callis Duffin K, Wong B, Horn EJ, Krueger GG. Psoriatic arthritis is a strong predictor of sleep interference in patients with psoriasis. J Am Acad Dermatol. 2009;60(4):604–608.
31. Vakil M, Park S, Broder A. The complex associations between obstructive sleep apnea and auto-immune disorders: a review. Med Hypotheses. 2018;110:138–143.
32. Machado-Pinto J, Diniz Mdos S, Bavoso NC. Psoriasis: new comorbidities. An Bras Dermatol. 2016;91(1):8–14.
33. Balci DD, Celik E, Genc S, Celik MM, Inan MU. Impaired pulmonary function in patients with psoriasis. Dermatology (Basel, Switzerland). 2016;232(6):664–667.
34. Li X, Kong L, Li F, et al. Association between psoriasis and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PloS One. 2015;10(12):e0145221.
35. Ungprasert P, Raksasuk S. Psoriasis and risk of incident chronic kidney disease and end-stage renal disease: a systematic review and meta-analysis. Int Urol Nephrol. 2018;50(7):1277–1283.
36. Chi C, Wang J, Chen Y, Wang S, et al. Risk of incident chronic kidney disease and end-stage renal disease in patients with psoriasis: a nationwide population-based cohort study. J Dermatol Sci. 2015 Jun;78(3):232–238
37. Yeung H, Takeshita J, Mehta NN, et al. Psoriasis severity and the prevalence of major medical comorbidity: a population-based study. JAMA Dermatol. 2013;149(10):1173–1179.
38. Molina-Leyva A, Jimenez-Moleon JJ, Naranjo-Sintes R, Ruiz-Carrascosa JC. Sexual dysfunction in psoriasis: a systematic review. J Eur Acad Dermatol Venereol. 2015;29(4):649–655.
39. Cabete J, Torres T, Vilarinho T, et al. Erectile dysfunction in psoriasis patients. Eur J Dermatol. 2014 ;24(4):482–486
40. Smolen JS, Landewe R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis. 2014;73(3):492–509
41. Roubille C, Richer V, Starnino T, et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Ann Rheum Dis. 2015;74(3):480–489.
42. Yang ZS, Lin NN, Li L, Li Y. The effect of TNF inhibitors on cardiovascular events in psoriasis and psoriatic arthritis: an updated meta-analysis. Clin Rev Allergy Immunol. 2016 Oct;51(2):240-7.
43. Levine B, Kalman J, Mayer L, et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. New Eng J Med. 1990;323(4):236–241.
44. Kadokami T, Frye C, Lemster B, et al. Anti-tumor necrosis factor-alpha antibody limits heart failure in a transgenic model. Circulation. 2001;104(10):1094–1097.
45. Chung ES, Packer M, Lo KH, et al. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):
3133–3140.
46. Rungapiromnan W, Yiu ZZN, Warren RB, et al. Impact of biologic therapies on risk of major adverse cardiovascular events in patients with psoriasis: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. 2017;176(4):890–901.
47. Ogdie A, Haynes K, Troxel AB, et al. Risk of mortality in patients with psoriatic arthritis, rheumatoid arthritis and psoriasis: a longitudinal cohort study. Ann Rheum Dis. 2014 Jan;73(1):
149-153.
48. Noe MH, Shin DB, Wan MT, Gelfand JM. Objective measures of psoriasis severity predict mortality: a prospective population-based cohort study. J Invest Dermatol. 2018;138(1):
228–230.
49. Alamdari HS, Gustafson CJ, Davis SA, et al. Psoriasis and cardiovascular screening rates in the United States. J Drugs Dermatol. 2013;12(1):
e14–19.
50. Manalo IF, Gilbert KE, Wu JJ. Survey of trends and gaps in dermatologists’ cardiovascular screening practices in psoriasis patients: areas still in need of improvement. J Am Acad Dermatol. 2015;73(5):872-874.
51. Rutter MK, Kane K, Lunt M, et al. Primary care-based screening for cardiovascular risk factors in patients with psoriasis. Br J Dermatol. 2016;175(2):348–356.
52. Micha R, Imamura F, Wyler von Ballmoos M, et al. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am J Cardiol. 2011;108(9):1362–1370.
53. Prodanovich S, Ma F, Taylor JR, Pezon C, Fasihi T, Kirsner RS. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. 2005;52(2):262–267.
54. Barnabe C, Martin BJ, Ghali WA. Systematic review and meta-analysis: anti-tumor necrosis factor alpha therapy and cardiovascular events in rheumatoid arthritis. Arthritis Care Res (Hoboken). 2011;63(4):522–529.
55. Wu JJ, Poon KY, Channual JC, Shen AY. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol. 2012;148(11):1244–1250.
56. Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti-inflammatory drugs: a Danish real-world cohort study. J Intern Med. 2013;273(2):197–204.
57. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory Tterapy with canakinumab for atherosclerotic disease. New Engl J Med. 2017;377(12):1119–1131.
58. Everett BM, Pradhan AD, Solomon DH, et al. Rationale and design of the Cardiovascular Inflammation Reduction trial: a test of the inflammatory hypothesis of atherothrombosis. Am Heart J. 2013;166(2):199–207.e115.
59. Bissonnette R, Harel F, Krueger JG, et al. TNF-alpha antagonist and vascular inflammation in patients with psoriasis vulgaris: a randomized placebo-controlled study. J Invest Dermatol. 2017;137(8):1638–1645.
60. Bissonnette R, Kerdel F, Naldi L, et al. Evaluation of risk of major adverse cardiovascular events with biologic therapy in patients with psoriasis. J Drugs Dermatol. 2017;16(10):1002–1013.
61. Chen YJ, Chen CC, Lin MW, et al. Increased risk of sexual dysfunction in male patients with psoriasis: a nationwide population-based follow-up study. J Sex Med. 2013;10(5):1212–1218.
62. Meeuwis KA, de Hullu JA, van de Nieuwenhof HP, et al. Quality of life and sexual health in patients with genital psoriasis. Br J Dermatol. 2011;164(6):1247–1255.
63. Meeuwis KA, van de Kerkhof PC, Massuger LF, et al. Patients’ experience of psoriasis in the genital area. Dermatology. 2012;224(3):271–276.
64. United States Centers for Disease Control and Prevention site. Recommended Immunization Schedule for Adults Aged 19 Years or Older, United States 2018. 6 Feb 2018. https://www.cdc.gov/vaccines/schedules/hcp/adult.html. Accessed 24 Sep 2018.
65. Marzano AV, Derlino F, Berti EF. Pathogenesis of psoriasis: focus on autoinflammation. Dermatopathology (Basel). 2018 Feb 2;5(1):14–15.
66. Elder JT. The quest for psoriasis autoantigens: genetics meets immunology in the melanocyte. J Invest Dermatol. 2017 Oct;137(10):2042–2045.
67. Chandran V. Genetics of psoriasis and psoriatic arthritis. Indian J Dermatol 2010;55(2):151–156.
68. Okada Y, Han B, Tsoi L, et al. Fine mapping of major histocompatibility complex associations in psoriasis and its clinical subtypes. Am J Hum Genet. 2014 Aug 7;95(2):162–72.
69. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645–653
70. Maxwell JR, Zhang Y, Brown WA, et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity. 2015;43(4):739–750.
71. Matos TR, O’Malley JT, Lowry EL, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. 2017;127(11):4031–4041.
72. National Psoriasis Foundation site. Statistics. Psoriatic Disease 2018. https://www.psoriasis.org/content/statistics. Accessed May 8, 2018.
73. Coates LC, Kavanaugh A, Mease PJ, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol. 2016 May;68(5):1060–1071
74. Husni ME, Mease PJ. Managing comorbid disease in patients with psoriatic arthritis. Curr Rheumatol Rep. 2010;12(4):281–287.
75. Kristensen LE, Jorgensen TS, Christensen R, et al. Societal costs and patients’ experience of health inequities before and after diagnosis of psoriatic arthritis: a Danish cohort study. Ann Rheum Dis. 2017;76(9):1495–1501.
76. Feldman SR, Zhao Y, Shi L, et al. Economic and comorbidity burden among moderate-to-severe psoriasis patients with comorbid psoriatic arthritis. Arthritis Care Res (Hoboken). 2015;67(5):708–717.
77. Evans C. Managed care aspects of psoriasis and psoriatic arthritis. Am J Manag Care. 2016;22(8 Suppl):s238–243.
78. Burgos-Pol R, Martinez-Sesmero JM, Ventura-Cerda JM, et al. The cost of psoriasis and psoriatic arthritis in 5 European countries: a systematic review. Actas Dermo-Sifiliograficas. 2016;107(7):577–590.
79. Ibrahim GH, Buch MH, Lawson C, et al. Evaluation of an existing screening tool for psoriatic arthritis in people with psoriasis and the development of a new instrument: the Psoriasis Epidemiology Screening Tool (PEST) questionnaire. Clin Exp Rheumatol. 2009;27(3):469–474
80. Reichert JM. Antibodies to watch in 2017. mAbs. 2017;9(2):167–181.
81. Papp K, Menter A, Strober B, et al. Efficacy and safety of brodalumab in subpopulations of patients with difficult-to-treat moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2015;72(3):436–439 e431.
82. Papp K, Pariser D, Catlin M, et al. Phase 2a randomised, double-blind, placebo-controlled, sequential dose-escalation study to evaluate the efficacy and safety of ASP015K, a novel Janus Kinase (JAK) inhibitor, in patients with moderate to severe psoriasis. Br J Dermatol. 2015;173(3):767–776.
83. Gossec L, Smolen JS, Ramiro S, et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis. 2016;75(3):
499–510.
84. Smolen JS, Braun J, Dougados M, et al. Treating spondyloarthritis, including ankylosing spondylitis and psoriatic arthritis, to target: recommendations of an international task force. Ann Rheum Dis. 2014;73(1):6–16.
85. Her M, Kavanaugh A. Alterations in immune function with biologic therapies for autoimmune disease. J Allergy Clin Immunol. 2016 ;137(1):
19–27.
86. Kavanaugh A, Husni ME, Harrison DD, et al. Safety and efficacy of intravenous golimumab in patients with active psoriatic arthritis: results through Week 24 of the GO-VIBRANT Study. Arthritis Rheumatol. 2017;69(11):
2151–2161.
87. Kavanaugh A, McInnes I, Mease P, et al. Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheumatol. 2009;60(4):976–986.
88. Fagerli KM, Kearsley-Fleet L, Watson KD, et al. Long-term persistence of TNF-inhibitor treatment in patients with psoriatic arthritis. Data from the British Society for Rheumatology Biologics Register. RMD Open. 2018;4(1):e000596.
89. Mease P, van der Heijde D, Landewe R, et al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann Rheum Dis. 2018;77(6):890-–97.
90. Mease PJ. Apremilast: a phosphodiesterase 4 inhibitor for the treatment of psoriatic arthritis. Rheumatol Ther. 2014;1(1):1–20.
91. Qu X, Zhang S, Tao L, Song Y. A meta-analysis of apremilast on psoriatic arthritis long-term assessment of clinical efficacy (PALACE). Exp Rev Clin Pharmacol. 2016;9(6):799–805.
92. Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. New Engl J Med. 2017;376(16):1551–1560.
93. Mease PJ, Kellner H, Morita A, et al. Efficacy and safety results from a Phase 2 trial of risankizumab, a selective IL-23p19 inhibitor, in patients with active psoriatic arthritis [abstract]. Arthritis Rheumatol. 2017; 69 (suppl 10). https://acrabstracts.org/abstract/efficacy-and-safety-results-from-a-phase-2-trial-of-risankizumab-a-selective-il-23p19-inhibitor-in-patients-with-active-psoriatic-arthritis/. Accessed September 27, 2018.
94. Bachelez H, van de Kerkhof PC, Strohal R, et al. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: a phase 3 randomised non-inferiority trial. Lancet. 2015;386(9993):552–561.
95. Mease P, Hall S, FitzGerald O, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. New Engl J Med. 2017;377(16):1537–1550.
96. Ogdie A, Schwartzman S, Eder L, et al. Comprehensive treatment of psoriatic arthritis: managing comorbidities and extraarticular manifestations. J Rheumatol. 2014;41(11):
2315–2322.
97. Shen J, Wong KT, Cheng IT, et al. Increased prevalence of coronary plaque in patients with psoriatic arthritis without prior diagnosis of coronary artery disease. Ann Rheum Dis. 2017;76(7):1237–1244.
98. Nickoloff BJ. Cracking the cytokine code in psoriasis. Nature Med. 2007;13(3):242–244.
99. Guttman-Yassky E, Krueger JG. Psoriasis: evolution of pathogenic concepts and new therapies through phases of translational research. Br J Dermatol. 2007;157(6):1103–1115.
100. Nickoloff BJ, Qin JZ, Nestle FO. Immunopathogenesis of psoriasis. Clin Rev Allergy Immunol. 2007;33(1-2):45–56.
101. Nickoloff BJ, Xin H, Nestle FO, Qin JZ. The cytokine and chemokine network in psoriasis. Clin Dermatol. 2007;25(6):568–573
102. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445(7130):866–873.
103. Drugs.com site. Development history and FDA approval process for Cimzia. Cimzia approval history 2018; https://www.drugs.com/history/cimzia.html. Accessed May 25, 2018.
104. McKnight W. CIMPASI-1 and -2 show certolizumab pegol benefits patients with severe plaque arthritis. Conference Coverage 2017; https://www.mdedge.com/edermatologynews/article/132706/psoriasis/cimpasi-1-and-2-show-certolizumab-pegol-benefits-patients. Accessed May 25, 2018.
105. Global Newswire site. CIMZIA (certolizumab pegol) Phase 3 trial meets co-primary endpoints in patients with moderate-to-severe chronic plaque psoriasis. Dermira press release. 8 Dec 2016. https://globenewswire.com/news-release/2016/12/08/895986/0/en/Second-CIMZIA-certolizumab-pegol-Phase-3-Trial-Meets-Co-primary-Efficacy-Endpoints-in-Patients-with-Moderate-to-Severe-Chronic-Plaque-Psoriasis.html. 27 Sep 2018.
106. Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–417.
107. A Deodhar, A Gottlieb, W-H Boehncke, et al. Efficacy and safety results of guselkumab in patients with active psoriatic arthritis over 56 weeks from a phase 2a, randomised, double-blind, placebo-controlled study. Abstract # OP0308. Presented at Annual European Congress of Rheumatology. Madrid, Spain: 14–16 Jun 2018. Ann Rheum Dis. 2018;77 (Suppl 2). https://ard.bmj.com/content/77/Suppl_2/201.1. Accessed 27 Sep 2018.
108. Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–288.
109. Bissonnette R, Luger T, Thaci D, et al. Secukinumab demonstrates high sustained efficacy and a favourable safety profile in patients with moderate-tosevere psoriasis through 5 years of treatment (SCULPTURE Extension Study). J Eur Acad Dermatol Venereol. 2018;32(9):1507–1514.
110. Blauvelt A, Gooderham M, Iversen L, et al. Efficacy and safety of ixekizumab for the treatment of moderate-to-severe plaque psoriasis: Results through 108 weeks of a randomized, controlled phase 3 clinical trial (UNCOVER-3). J Am Acad Dermatol. 2017;77(5):855–862.
111. Paul C, Griffiths CEM, van de Kerkhof PCM, et al. AIxekizumab provides superior efficacy compared to ustekinumab over 52-weeks of treatment: results from IXORA-S, a phase 3 study. J Am Acad Dermatol. 2018 Jun 30. pii: S0190-9622(18)32195-9. doi: 10.1016/j.jaad.2018.06.039. [Epub ahead of print].
112. Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. New Engl J Med. 2015;373(14):
1318–1328.
113. United States Food and Drug Administration site. FDA Briefing Document. Background Package for BLA 761032 Siliq (brodalumab) injection, 210 mg/1.5 ml. Dermatologic and Ophthalmic Drugs Advisory Committee. meeting. July 19, 2016. https://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/ drugs/dermatologicandophthalmic drugsadvisorycommittee/ucm511357.pdf. Accessed 27 Sep 2018.
114. Gupta MA, Schork NJ, Gupta AK, et al. Suicidal ideation in psoriasis. Int J Dermatol. 1993;32(3):188–190.
115. Lonnberg AS, Zachariae C, Skov L. Targeting of interleukin-17 in the treatment of psoriasis. Clin Cosmet Investig Dermatol. 2014;7:251-259.
116. Glatt S, Helmer E, Haier B, et al. First-in-human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL-17A and IL-17F, in mild psoriasis. Br J Clin Pharmacol. 2017;83(5):991-1001.
117. Kircik L, Lebwohl MG, Del Rosso JQ, et al. Clinical study results of desoximetasone spray, 0.25% in moderate to severe plaque psoriasis. J Drugs Dermatol. 2013;12(12):1404–1410.
118. Patel P, Schmieder S, Krishnamurthy K. Research techniques made simple: drug delivery techniques, part 2: commonly used techniques to assess topical drug bioavailability. J Invest Dermatol. 2016;136(5):e43–49.
119. Fowler JF, Jr., Hebert AA, Sugarman J. DFD-01, a novel medium potency betamethasone dipropionate 0.05% emollient spray, demonstrates similar efficacy to augmented betamethasone dipropionate 0.05% lotion for the treatment of moderate plaque psoriasis. J Drugs Dermatol. 2016;15(2):154–162.
120. Koo J, Tyring S, Werschler WP, et al. Superior efficacy of calcipotriene and betamethasone dipropionate aerosol foam versus ointment in patients with psoriasis vulgaris: a randomized phase II study. J Dermatolog Treat. 2016;27(2):120–127.
121. Torsekar R, Gautam MM. Topical therapies in psoriasis. Indian Dermatol Online J. 2017;8(4):
235–245
122. Chandraratna R. Tazarotene: the first receptor-selective topical retinoid for the treatment of psoriasis. J Am Acad Dermatol. 1997;37(2):
S12–S17.
123. Gold LS, Lebwohl MG, Sugarman JL, et al. Safety and efficacy of a halobetasol/tazarotene fixed combination in the treatment of moderate-to-severe plaque psoriasis: results of two Phase 3 randomized controlled trials. J Am Acad Dermatol. 2018 Aug;79(2):287-293
124. Bissonnette R, Vasist LS, Bullman JN, et al. Systemic pharmacokinetics, safety, and preliminary efficacy of topical AhR agonist tapinarof: results of a Phase 1 study. Clin Pharmacol Drug Dev. 2018;7(5):524–531.
125. Smith SH, Jayawickreme C, Rickard DJ, et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J Invest Dermatol. 2017;137(10):2110–2119.
126. A dose-finding study of GSK2894512 cream in subjects with plaque psoriasis. 2015. ClinicalTrials.gov Identifier: NCT02564042. https://clinicaltrials.gov/ct2/show/. Accessed 27 Sep 2018. NCT02564042?term= GSK2894512&cond= plaque+ psoriasis&rank=1. Accessed 27 May 2018.
127. Punwani N, Burn T, Scherle P, et al. Downmodulation of key inflammatory cell markers with a topical Janus kinase 1/2 inhibitor. Br J Dermatol. 2015;173(4):989–997.
FUNDING: This supplement was funded by Sun Pharmaceutical Industries Inc. in Princeton, New Jersey.
FINANCIAL DISCLOSURES:
Dr. Blauvelt has served as a scientific adviser, clinical study investigator, or paid speaker for AbbVie, Aclaris, Akros, Allergan, Almirall, Amgen, Boehringer Ingelheim, Celgene, Dermavant, Dermira, Inc., Eli Lilly and Company, Galderma, Genentech/Roche, GlaxoSmithKline, Janssen, Leo, Meiji, Merck Sharp & Dohme, Novartis, Pfizer, Purdue Pharma, Regeneron, Revance, Sandoz, Sanofi Genzyme, Sienna Pharmaceuticals, Sun Pharma, UCB Pharma, Valeant, and Vidac, Janssen, Regeneron, and Sanofi Genzyme.
Dr. Gelfand has received honoraria, research grants (indirectly, to the Trustees of the University of Pennsylvania), or payment for continuing medical education work from AbbVie, BMS, Boehringer Ingelheim, Celgene, GSK, Janssen Biologics, Lilly, Novartis Corp, Ortho Dermatologics, Pfizer Inc., Regeneron, Sanofi, and UCB (DSMB).
Dr. Kavanaugh has conducted research studies for AbbVie, Amgen, Astra-Zeneca, BMS, Celgene, Corrona , Galapagos/Gilead, Genentech/Roche, ITN, Janssen, LCTC, LIAI, NIH, Novartis, Pfizer, Regeneron/Sanofi, TREG, and UCB.
Dr. Leonardi has served as a consultant, speaker, advisory board member, or investigator for Abbvie, Allergan, Amgen, Celgene, Coherus, Dermira, Eli-Lilly, Galderma, Glaxo Smith Kline, Janssen, Leo, Merck, Merck-Serono, Novartis, Pfizer, Sandoz, Sienna, UCB, and Vitae.
Ms. LeQuang has no conflicts of interest relative to the content of this supplement.
Dr. Martin has served as a speaker, consultant, or advisory board member for Aclaris, Aqua, Celgene, DUSA/Sun Pharma, Janssen, Pfizer, Ortho/Valeant, UCB, Aclaris, Celgene, Pfizer, and UCB.
Dr. Stein Gold has served as a speaker, consultant, or advisory board member for Leo Pharma, Mayne Pharma, Pfizer Inc., Taro, and Valeant.
Dr. Strober has been a consultant, investigator (did not receive direct payments), or a scientific director or has received grant support (indirectly, to the University of Connecticut for Fellowship Program) from AbbVie, Almirall, Amgen, Boehringer Ingelheim, Bristol-Myers-Squibb, CORRONA Psoriasis Registry, Celgene, Dermavant, Dermira, Galderma, GlaxoSmithKline, Janssen, Leo, Eli Lilly, Leo, Medac, Meiji Seika Pharma, Merck, Menlo Therapeutics, National Psoriasis Foundation, Novartis, Ortho Dermatologics/Valeant, Pfizer, Regeneron, Sanofi-Genzyme, Sebela Pharmaceuticals, Sirtris, Sun Pharma, and UCB Pharma.