J Clin Aesthet Dermatol. 2019;12(11):23–26
 by Sabeen Yaqub, MD; Shelly A. Stepenaskie, MD; Fatemeh Jafari Farshami, MD; Wilmer L. Sibbitt Jr., MD; Monthida Fangtham, MD; N. Suzanne Emil, MD; and Arthur D. Bankhurst, MD
by Sabeen Yaqub, MD; Shelly A. Stepenaskie, MD; Fatemeh Jafari Farshami, MD; Wilmer L. Sibbitt Jr., MD; Monthida Fangtham, MD; N. Suzanne Emil, MD; and Arthur D. Bankhurst, MD
Drs. Yaqub, Farshami, Sibbitt, Fangtham, Emil, and Bankhurst are with the Department of Internal Medicine, Division of Rheumatology, and School of Medicine at the University of New Mexico Health Sciences Center in Albuquerque, New Mexico. Dr. Stepenaskie is with the Departments of Dermatology and Pathology at the University of New Mexico Health Sciences Center in Albuquerque, New Mexico.
FUNDING: No funding was provided for this study.
DISCLOSURES: The authors have no conflicts of interest relevant to the content of this article.
ABSTRACT: Painful, palpable purpura usually indicate underlying vasculitis. We report a case of systemic vasculitis treated with immunosuppression that developed painful, vasculitis-like purpuric lesions that progressed rapidly to fulminant Kaposi sarcoma (KS). These purpuric, tumorous lesions resolved completely following the suspension of immunosuppression; however, without immunosuppression, the underlying autoimmunity recurred. This case highlights the potential for early KS to present as a vasculitis mimic or pseudovasculitis that clinicians should keep in mind when purpuric, vasculitis-like lesions develop in an immunosuppressed patient with vasculitis. It is important to recognize these pseudovasculitis lesions as KS rather than recurrent vasculitis so that immunosuppression can be withdrawn.
KEYWORDS: Immunosuppression, Kaposi sarcoma, purpura, vasculitis
Pseudovasculitis or vasculitis mimics are diseases that can be confused with autoimmune vasculitis.1 Typical vasculitis mimics include atherosclerosis, thromboembolic disease, atrial myxoma, cholesterol emboli syndrome, hypercoagulability states, congenital disorders of collagen, vasospastic disease, drug-induced vascular disease, calciphylaxis, and sometimes platelet disorders and hemorrhagic states.1,2 However, Kaposi sarcoma (KS) is rarely listed as a vasculitis mimic.3 KS is a malignant vascular tumor that can develop in association with congenital or acquired immunodeficiency disease, especially in cases of untreated human immunodeficiency virus (HIV) and more rarely in those receiving immunosuppressive therapy.4 We report the case of a 64-year-old woman with a diagnosis of microscopic polyangiitis who was treated with standard immunosuppression and developed painful, vasculitis-like purpuric skin lesions that, on biopsy, were proven to be KS.
Case Presentation
A 64-year-old woman with a past medical history of diabetes mellitus and hypertension presented with a three-month history of shortness of breath on exertion and chronic intermittent hemoptysis. An examination demonstrated a vasculitic palpable purpuric rash in the lower extremities and synovitis in the metacarpophalangeal and proximal interphalangeal joints. Extensive laboratory evaluation revealed elevated perinuclear antineutrophil cytoplasmic antibody (p-ANCA) (1:640 titer) and anti-myeloperoxidase (MPO) antibody (>8IU). A computed tomography (CT) scan of the chest was concerning for bronchiolitis obliterans with organizing pneumonia (BOOP), which was confirmed on lung biopsy. Based on the observation of vasculitic skin lesions, lung involvement, and characteristic serologies with positive MPO and p-ANCA results, the patient was diagnosed with systemic vasculitis microscopic polyangiitis and initially started on high-dose corticosteroids. Within a few days, the skin lesions, infiltrates, and hemoptysis had completely resolved. However, as the corticosteroids were tapered, the vasculitic rash, pulmonary lesions, and hemoptysis recurred and pulse corticosteroids and intravenous cyclophosphamide were initiated. Cyclophosphamide initially resulted in a complete resolution of hemoptysis and vasculitic skin lesions. After three cyclophosphamide doses, the corticosteroids were tapered and azathioprine maintenance therapy was initiated at 150mg daily.
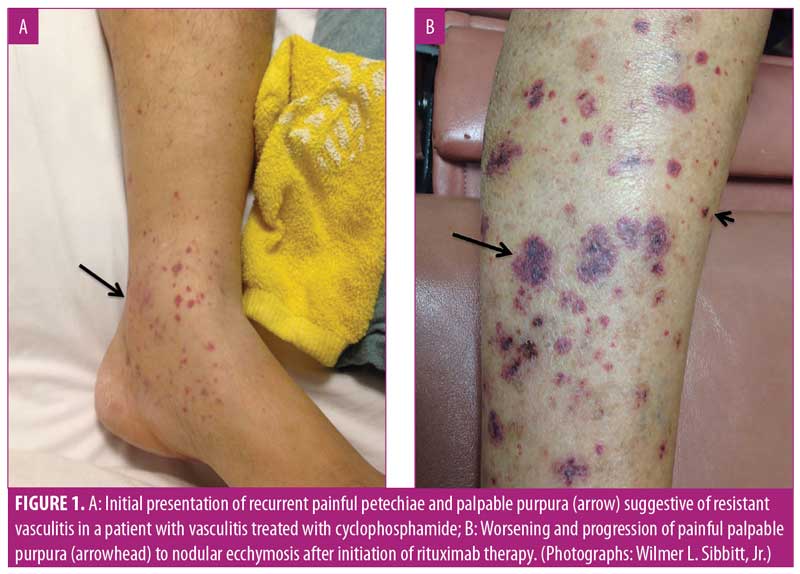
After five months on azathioprine, the patient developed new painful, purpuric, vasculitis-like lesions on the lower extremities that appeared clinically identical to her initial vasculitis skin lesions (Figure 1A). Because of concerns of a vasculitic flare, the azathioprine dose was increased to 200mg and prednisone was increased to 60mg per day. Two months later, the patient complained of considerable pain and worsening purpuric rash that had also spread to her hands and feet. The patient stated that these new lesions were identical to those seen when she first developed vasculitis. Her erythrocyte sedimentation rate (ESR) and C-reactive protein level (CRP) were markedly elevated; however, ANCA and MPO tests were negative. With concerns regarding a vasculitic flare, intravenous rituximab (1,000mg) was initiated. Two weeks after the first rituximab dose, new perioral lesions developed and the rash rapidly progressed (Figure 1B). Based on the morphology of the exuberant skin lesions and the negative vasculitis serologies, a clinical diagnosis of KS was made, all immunosuppression was discontinued, and a skin biopsy was obtained. The skin biopsy results were also indicative of KS, with sheets of spindle cells, slit-like blood vessels, extravasated red blood cells, and hemosiderin deposits (Figure 2). An immunohistochemical stain for human herpes virus-8 (HHV-8) demonstrated the presence of HHV-8 within the spindled endothelial cells (Figure 3). The patient was negative for HIV serologically and by polymerase chain reaction amplification. Despite the discontinuation of immunosuppression, the cutaneous lesions rapidly progressed to fungating, ulcerative, and elevated lesions (Figure 4A). CT demonstrated tumorous lesions in the lungs and liver consistent with Stage 3 KS and she was evaluated by oncology. After the cessation of immunosuppression, the lung, liver, and skin lesions resolved completely without further treatment over the subsequent four months (Figure 4B). However, at eight months, the patient began to complain of arthralgias, and the p-ANCA (1:160 titer) and MPO antibody (1.8 IU) findings again were positive, suggesting recurrent vasculitis, although no cough, hemoptysis, lung lesions, or skin lesions were noted. Because the ESR, CRP, and urinalysis results were normal, despite the conversion to positive ANCA serologies, and given the recent history of KS, it was deemed most prudent to observe the patient and only initiate immunosuppression if a true vasculitic recurrence were observed.



Discussion
The skin lesions of KS are typically characterized by purple, red, or brown blotches or tumors on the skin, similar to those shown in Figure 1B and Figure 4A.3–5 As recently reviewed by Marušic and Billings,5 KS demonstrates an abnormally enhanced vascularity (Figure 2); thus, because of this superficial vascularity, at certain stages, KS can be confused with other tumors and vascular processes.5 As with the palpable purpura of vasculitis, the dark color and hemorrhagic nature of the skin lesions of KS is due to the enhanced vascularity of the disease, extravasation of red blood cells, and the deposition of hemosiderin in the dermis (Figure 2). Although the initial rash of KS in the present case appeared as small, painful, vascular purpura that mimicked vasculitic purpura clinically (Figure 1A), the lesions rapidly progressed to show an atypical morphology (Figure 1B) and then expanded with further immunosuppression (Figure 4A). The lesions in Figure 1A and perhaps Figure 1B could reasonably be confused with vasculitis; however, the extremely atypical nodular, fungating, and violaceous lesions in Figure 4A are classic for KS. Thus, at this point, it was crucial to rethink the diagnosis, stop immunosuppression, and biopsy the lesions once it was confirmed there were atypical skin lesions and a lack of clinical improvement with immunosuppression.
As reviewed in depth by Carlson et al,1 pseudovasculitis or vasculitis mimics are diseases that can be confused with true vasculitis. Typical vasculitis mimics include malignancy, atrial myxoma, atherosclerosis, thromboembolic disease, hypercoagulability states, congenital disorders of collagen, calciphylaxis, vasospastic disease, drug-induced vascular disease (e.g., caused by adrenergic agents, cocaine, levamisole, ergotism), thromboangiitis obliterans, infections (e.g., sepsis, rickettsial disease, viral syndromes, bacterial endocarditis), and sometimes platelet disorders and hemorrhagic states, as well as many other conditions.1,2 The present report details a case of systemic vasculitis with ANCA/MPO positivity, skin vasculitis, arthritis, hemoptysis, and BOOP-like pulmonary lesions that, with immunosuppression, developed KS cutaneous palpable purpura that, initially, clinically mimicked recurrent cutaneous vasculitis. Although KS is a well-known complication of immunosuppression among autoimmune diseases, there is only one prior report of KS as a vasculitis mimic.3
As recently reviewed by Vangipuram et al,4 KS is generally classified as either epidemic (HIV-associated), classic (Mediterranean), endemic (African), or iatrogenic (transplant-related).4 Epidemic KS, usually associated with HHV-8, has become increasingly rare in industrialized countries as a result of efficient HIV detection and effective antiviral therapy, yet still occurs frequently in other parts of the world.6 Classic KS occurs often in older people of Mediterranean, Eastern European, or Middle Eastern heritage, and generally, the disease can be more self limited and localized as a result of a less impaired immune system, but also appears to be associated with HHV-8.7 Endemic KS caused by HHV-8 occurs most commonly in people living in Equatorial Africa, where primary HHV-8 infection is more common than the rest of the world; recently increased case numbers have also been associated with untreated HIV infections in Africa.8
The present case would be regarded as that of iatrogenic KS in an immunosuppressed autoimmune patient. Iatrogenic KS has been most commonly reported in transplant patients or autoimmune patients who are intensely immunosuppressed, and the cutaneous lesions, as seen in this case, often resolve with cessation of immunosuppression.9,10 There are multiple reported cases of KS in autoimmune patients that developed following immunosuppression.9,11–16 As in this case, most of known the iatrogenic KS cases are associated with endemic HHV-8 infection (Figure 3).9 However, there is one prior reported case of KS being a vasculitis mimic (pseudovasculitis); thus, early KS should be included on the lists of vasculitis mimics and pseudovasculitis.3 Of note, KS in immunosuppressed autoimmune patients can constitute a difficult problem to manage in the long term, in that, after the immunosuppression is withdrawn and the KS resolves, the vasculitis and autoimmune syndrome can recur.
Conclusion
KS is a rare iatrogenic complication that can develop in immunosuppressed patients with vasculitis, creating vascular purpuric lesions mimicking recurrent vasculitis—a true vasculitis mimic (pseudovasculitis). It is important to recognize these pseudovasculitis lesions as KS rather than recurrent vasculitis so that immunosuppression can be withdrawn instead of increased. It is challenging to treat ANCA vasculitis in such patients, in that, with immunosuppression, KS can develop and, when the immunosuppression is withdrawn, the underlying systemic vasculitis can recur.
References
- Carlson JA, Chen KR. Cutaneous pseudovasculitis. Am J Dermatopathol. 2007;29(1):44–55.
- Grau R. Pseudovasculitis: mechanisms of vascular injury and clinical spectrum. Curr Rheumatol Rep. 2002;4(1):83–89.
- Kaur S, Erm T. Multifocal Kaposi’s sarcoma mimicking allergic vasculitis. Acta Derm Venereol. 2004;84(2):156–157.
- Vangipuram R, Tyring SK. Epidemiology of Kaposi sarcoma: review and description of the nonepidemic variant. Int J Dermatol. 2019;58(5):538–542.
- Marušic Z, Billings SD. Histopathology of spindle cell vascular tumors. Surg Pathol Clin. 2017;10(2):345–366.
- Chang E, Mapakshi SR, Mbang P, et al. The impact of protease inhibitors on HIV-associated Kaposi sarcoma incidence: a systematic review. J Acquir Immune Defic Syndr. 2018;79(2):141–148.
- Di Lorenzo G. Update on classic Kaposi sarcoma therapy: new look at an old disease. Crit Rev Oncol Hematol. 2008;68(3):242–249.
- Simonart T. Role of environmental factors in the pathogenesis of classic and African-endemic Kaposi sarcoma. Cancer Lett. 2006;244(1):1–7.
- Périer A, Savey L, Marcelin AG, et al. De novo human herpesvirus 8 tumors induced by rituximab in autoimmune or inflammatory systemic diseases. Arthritis Rheumatol. 2017;69(11): 2241–2246.
- Hassan G, Khalaf H, Mourad W. Dermatologic complications after liver transplantation: a single-center experience. Transplant Proc. 2007;39(4):1190–1194.
- Kiliç DT, Özkan SK, Canbakan B, Dündar N. Kaposi’s sarcoma concurrent with granulomatosis polyangiitis. Eur J Rheumatol. 2016;3(2):85–86.
- Berti A, Scotti R, Rizzo N, et al. Kaposi’s sarcoma in a patient with eosinophilic granulomatosis with polyangiitis while taking mycophenolate mofetil. J Allergy Clin Immunol Pract. 2015;3(3):431–432.
- Fatma LB, Rais L, Mebazza A, et al. Kaposi’s sarcoma with HHV8 infection and ANCA-associated vasculitis in a hemodialysis patient. Saudi J Kidney Dis Transpl. 2013;24(6):1199–1202.
- Bouattar T, Kazmouhi L, Alhamany Z, et al. Kaposi’s sarcoma following immunosuppressive therapy for vasculitis. Saudi J Kidney Dis Transpl. 2011;22(2):319–323.
- Louthrenoo W, Kasitanon N, Mahanuphab P, et al. Kaposi’s sarcoma in rheumatic diseases. Semin Arthritis Rheum. 2003;32(5):326–333.
- Hoff M, Rodevand E. Development of multiple malignancies after immunosuppression in a patient with Wegener’s granulomatosis. Rheumatol Int. 2005;25(3):238–240.
- Deschenes I, Dion L, Beauchesne C, de Brum-Fernandes A. Kaposi’s sarcoma following immune suppressive therapy for Wegener’s granulomatosis. J Rheumatol. 2003;30(3):622–624.
- Erban SB, Sokas RK. Kaposi’s sarcoma in an elderly man with Wegener’s granulomatosis treated with cyclophosphamide and corticosteroids. Arch Intern Med. 1988;148(5):1201–1203.