J Clin Aesthet Dermatol. 2025;18(3–4 Suppl 1):16–23.
by Matthew Zirwas, MD; Cynthia Trickett, PA-C, MPAS; Joe Gorelick, MSN, FNP-C; Kejia Wang, MA, PhD; Keith Wittstock, PharmD, MBA; Chaya Rosenberg, DNP, RN, CNL, RD; and Douglas DiRuggiero, DMSc, PA-C
Dr. Zirwas is with DOCS Dermatology in Bexley, Ohio. Ms. Trickett is with North Dallas Dermatology Associates in Dallas, Texas. Mr. Gorelick is with the California Skin Institute in San Jose and Los Gatos, California. Ms. Wang, Mr. Wittstock, and Ms. Rosenberg are with Bristol Myers Squibb in Princeton, New Jersey. Mr. DiRuggiero is with the Skin Cancer and Cosmetic Dermatology Center in Rome, Georgia.
Funding: This review was sponsored by Bristol Myers Squibb. Writing and editorial assistance was provided by Ann Marie Fitzmaurice, PhD, of Peloton Advantage, LLC, an OPEN Health company, funded by Bristol Myers Squibb.
Disclosures: Dr. Zirwas has served as a consultant, investigator, and/or speaker for AbbVie, Acrotech, Advanced Derm Solutions, Aldeyra, All Free Clear/Sun, Amgen, AnaptysBio, Apogee, Arcutis, Bausch and Lomb, Beiersdorf, Biocon, Bristol Myers Squibb, Cara Therapeutics, Castle Biosciences, Dermavant, Edessa Biotech, Evelo, Galderma, Google, Incyte, Janssen, L’Oreal, Leo Pharma, Level-Ex, Lilly, LUUM, Meta, Nimbus, Novan, Novartis, Pfizer, Q32 Bio, Regeneron, Sanofi, Sun Pharma, Supernus, Takeda, Trevi, Trifecta, UCB, and Verrica. Ms. Trickett has served as a speaker and consultant for AbbVie, Amgen, Bristol Myers Squibb, Incyte, Janssen, Journey Medical, Leo Pharma, Lilly, Ortho, Pfizer, Regeneron, Sanofi Genzyme, Sun Pharma, and UCB. Mr. Gorelick has served as a consultant and/or speaker for AbbVie, Amgen, Arcutis, Beiersdorf, Bristol Myers Squibb, Dermavant, Galderma, Incyte, Janssen, Leo Pharma, Lilly, Lyceum, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharma, UCB, and Verrica. Ms. Wang, Mr. Wittstock, and Ms. Rosenberg are employees of and shareholders in Bristol Myers Squibb. Mr. DiRuggiero has served as a speaker and consultant for AbbVie, Amgen, Arcutis, Bristol Myers Squibb, EPI Health, Incyte, Janssen, Leo Pharma, Lilly, Novartis, Pfizer, Sanofi Regeneron, and UCB.
ABSTRACT: Clinical trials are designed to evaluate the efficacy and safety of new drugs. However, greater focus is often placed on efficacy rather than safety. This review article discusses the fundamentals involved in evaluating the safety of a new drug. In addition, the principal challenges involved in the collection, analysis, reporting, and interpretation of safety data in clinical trials are described using relevant examples. These challenges include the fact that clinical trials are generally limited in size and duration, exclude high-risk populations, and have limited statistical power to detect rare but potentially serious adverse events (AEs) that might occur in real-world situations. Reporting of safety data across clinical trials is also inconsistent. A thorough understanding of the interpretation of safety data, especially the appropriate use of exposure-adjusted incidence rates (EAIRs) in relation to AEs, as well as the importance of comparing rates to those reported in the general population and in patients with psoriasis, is vital for making a well-informed assessment of the safety of a new drug. The information provided in this article could be useful to healthcare providers who must evaluate a large volume of safety data when providing evidence-based treatment suggestions and recommendations to their patients. Keywords: Adverse events, exposure-adjusted incidence rates, laboratory parameters, randomized controlled trials, safety
Introduction
Clinical trials are designed to evaluate the efficacy and safety of new drugs.1 However, greater focus is often placed on efficacy in many clinical trials, and only selective reporting of safety data can occur. Statistical approaches used to analyze efficacy data tend to be more rigorous than those applied to safety data.1 Safety data are also frequently presented in tabular rather than graphical form, which can hinder the interpretation of results.2 Therefore, inadequacies exist in the capture, analysis, and reporting of safety data, which can preclude a thorough evaluation of the risk-benefit profile of a new drug.
This review article is the second in a series from our group that discusses how variations in study design and analytical methodologies affect interpretation of clinical outcomes. The first review article in this series focused on clinical efficacy.3 The objective of this review is to educate healthcare providers about safety analyses commonly used in psoriasis clinical trials. Challenges inherent in the collection, analysis, and reporting of safety data in clinical trials are discussed. This information could be useful to healthcare providers who must evaluate a large volume of safety data based on varying study designs and analytical methodologies when attempting to provide evidence-based treatment recommendations to their patients.
Safety analyses in clinical trials
Safety analyses are conducted in clinical trials to allow thorough understanding of the safety profile of a new drug (Figure 1).2 Safety analyses help to determine whether the new drug poses any major safety concerns, identify relevant safety signals and associated risk factors as early as possible, prevent or mitigate additional safety issues where feasible, and reduce the attrition rate due to safety and/or tolerability concerns.4 Comprehensive safety analyses also minimize the likelihood of the post-approval emergence of previously unidentified safety signals and help to identify areas requiring additional safety monitoring.4

Safety is evaluated throughout the clinical development process. In early phase trials, safety is the primary focus, whereas efficacy becomes increasingly important in later phases (Figure 2).1

Phase 1 and 2 trials explore safety and determine the maximum tolerated dose of the drug, human pharmacokinetics and pharmacodynamics, and the potential for drug-drug interactions.1 Phase 1 and 2 trials both evaluate predictable pharmacologic-based toxicities, including liver toxicity (ie, elevations in liver enzyme levels such as alanine aminotransferase and aspartate aminotransferase), and hematologic suppression (eg, reductions in hemoglobin levels).1,5
Phase 3 trials seek to confirm and establish dosing regimens and to provide data about dose titrations after the initial primary outcome time point has been reached. In addition, Phase 3 trials are designed to confirm that toxicities observed in Phase 1 and 2 trials are rare and to identify additional unexpected events. Phase 3 trials can include a placebo arm and/or a comparator arm.1
Phase 4 trials assess long-term safety in a real-world setting, including the potential for less common events that do not occur in Phase 3 trials.1 Unlike Phase 3 trials, which have strict eligibility criteria, Phase 4 trials are conducted in a broad patient population with diverse patient demographics and clinical characteristics, reflecting how the new medication will be used in patients’ daily lives.1 Both Phase 3 and Phase 4 trials seek to identify an increased risk of idiosyncratic events associated with the study drug, such as an increased risk of infections in patients treated with immunosuppressive agents and an increased risk of deep vein thrombosis in patients treated with agents that promote blood clot formation.1
Adverse events. Definitions and grades. An adverse event (AE) is any untoward or unfavorable medical occurrence in a patient, including any abnormal sign (eg, abnormal physical examination or laboratory finding), symptom, or disease that is temporally associated with the patient’s involvement in the clinical trial, whether or not the event is considered related to participation in the trial.6 A serious adverse event (SAE) is an AE that results in death, is life-threatening or places the patient at immediate risk for death, requires or prolongs hospitalization, causes persistent or significant disability or incapacity, or results in abortion (spontaneous or voluntary), congenital anomalies, or birth defects.7
Adverse events are classified into categories or grades according to clinical severity using the Common Terminology Criteria for Adverse Events (CTCAE; Table 1).6 Notably, severity is not synonymous with seriousness. Severity is a measure of intensity of symptoms or negative impact on physical functioning and, thus, is often unrelated to seriousness. For example, mild chest pain that does not limit ability to perform normal daily activities might be serious if it results in overnight hospitalization to rule out a myocardial infarction. In contrast, a severe migraine headache that prevents performing any normal daily activities for 10 hours would not be serious unless it resulted in hospitalization. Some patients might also experience multiple events, which can be interrelated (eg, hospitalization for chest pain followed by a stent procedure, headache, fatigue, nausea). The severity grade and/or seriousness might need to be determined separately for each event, especially in situations where events are not clearly interrelated.

Treatment relatedness.The potential relationship of an AE to study treatment is usually assessed by the investigator. The National Institute on Aging has the following guideline that is commonly used to categorize events:
Definitely related: The AE is clearly related to study treatment; the event follows a reasonable temporal sequence from administration of treatment or follows a known or expected response pattern to treatment that is confirmed by improvement on stopping and reappearance on repeated exposure, and that could not be reasonably explained by the known characteristics of the patient’s clinical state.8 For example, a patient develops anemia after taking a drug known to suppress hematopoiesis; the anemia resolves after stopping the drug and recurs after restarting the drug.
Possibly related: An AE follows a reasonable temporal sequence from administration of treatment or follows a known or expected response pattern to treatment that could also readily have been produced by various other factors.8 For example, a patient develops a sinus infection after taking a drug known to cause immunosuppression. However, since this patient also has a history of sinusitis, the relationship to the study drug cannot be clearly established.
Not related: The AE is clearly not related to treatment; another cause is more plausible, and/or a clinically plausible temporal sequence is inconsistent with the onset of the event, and/or a causal relationship is considered biologically implausible.8 For example, a patient trips and breaks their wrist after using a topical steroid for one day on their extensor elbow. Similarly, a patient experiences an AE resulting in hospitalization 1.5 years after receiving a single dose of a new drug; although this AE is classified as serious, the temporal sequence of events suggests that this AE is not treatment related.
Certain AEs might undergo adjudication, whereby the study sponsor requests additional source documentation from the investigator to better evaluate the event and determine treatment-relatedness. Specific protocol-defined AEs, generally identified based on the mechanism of action of the drug or prior clinical experience, such as infections, cardiovascular events, malignancies, and/or suicidal ideation and behavior, might also be adjudicated by independent, subspecialty expert adjudicators blinded to treatment assignment.9–12 Involvement of subspecialty expert adjudicators can confer added rigor to the assessment of treatment-relatedness. In situations where a safety event has been adjudicated, clinicians with expertise related to specific safety events make a determination whether the event is related to the study drug or not. For example, a 60-year-old man developed deep vein thrombosis two years after starting a new drug for psoriasis. After reviewing the patient’s medical records, subspecialty adjudicators with expertise in vascular surgery, hematology, cardiology, and interventional radiology determined that the deep vein thrombosis was not related to treatment because the patient had several risk factors for this event, including smoking history, sedentary lifestyle, obesity, heart failure, and a history of pulmonary embolism.
Boxed warnings and prescribing information. Regulatory authorities such as the US Food and Drug Administration (FDA) occasionally require pharmaceutical companies to add a boxed warning (formerly known as a black box warning) to the drug prescribing information. A boxed warning is the strongest warning that the FDA issues, and it signifies that, in the opinion of the FDA, the drug carries a significant risk of preventable, serious, or even life-threatening AEs.13 Warnings might also be issued to highlight dosing issues, increased drug-drug interaction potential, and monitoring requirements. Boxed warnings are supposed to be based on detection of an elevated risk for a specific event in pivotal clinical trials, or safety concerns identified by the Adverse Event Reporting System operated by the FDA, which oversees postmarketing safety evaluations.13 However, the exact threshold that must be met to achieve boxed warning status remains somewhat nebulous. Boxed warnings can be applied as drug class warnings, whereby all drugs within a specific class receive a boxed warning based on safety concerns identified for older agents in this class. As an example, the prescribing information for Janus kinase (JAK) 1, 2, and 3 inhibitors used to treat inflammatory diseases includes a boxed warning regarding an increased risk of major adverse cardiovascular events (MACE), venous thromboembolic events, serious infections, malignancies, and death based on the results of clinical trials conducted in patients with rheumatoid arthritis.14–16 Although it is still in question whether newer generation JAK 1, 2, and 3 inhibitors or topical JAK inhibitors carry similar risks, each drug in this class carries the boxed warning.17 Similarly, IL-23, IL-12/IL-23, and IL-17 inhibitors require monitoring for tuberculosis before initiating treatment because of an earlier boxed warning requirement for tuberculosis monitoring before and during treatment with tumor necrosis factor inhibitors.18–28 As a result of these caveats, it is important for the reader to consider the specific verbiage used in the prescribing information to determine whether a specific AE has been detected in a clinical trial of a new drug or whether this drug poses an evidence-based increased risk for a specific AE (eg, ‘cancer has been detected with drug X’ vs. ‘cancer has been reported at a higher incidence rate with drug X vs. placebo or an active comparator’).
Various challenges are associated with the evaluation of safety data in clinical trials, including those related to study design, data collection, data analysis, and interpretation of results (Table 2).2,4,29 Most clinical trials are designed with strict eligibility criteria that might exclude patients with comorbid conditions and/or patients using concomitant medications; the result of these approaches is to limit the generalizability of safety findings in real-world populations.4 For instance, clinical trials that exclude patients with a history of depression might report a low incidence of depression during the trial; however, given how common this condition is in the general population, the incidence of depression might be statistically higher in patients receiving treatment with the new drug in a real-world situation. Although some safety events can be anticipated based on the mechanism of action of the drug and the results of earlier clinical trials, rarer events might only be detectable when large numbers of patients use the drug after it has received FDA approval, even when appropriate safety analyses have been performed.30 Clinical trials are usually powered to assess efficacy rather than safety; because of the limited size of the study population and duration of treatment (often in the range of 1,000–2,000 patients treated for one year) and the rarity of some safety events, clinical trials are generally underpowered to perform quantitative safety assessments across treatment arms.30
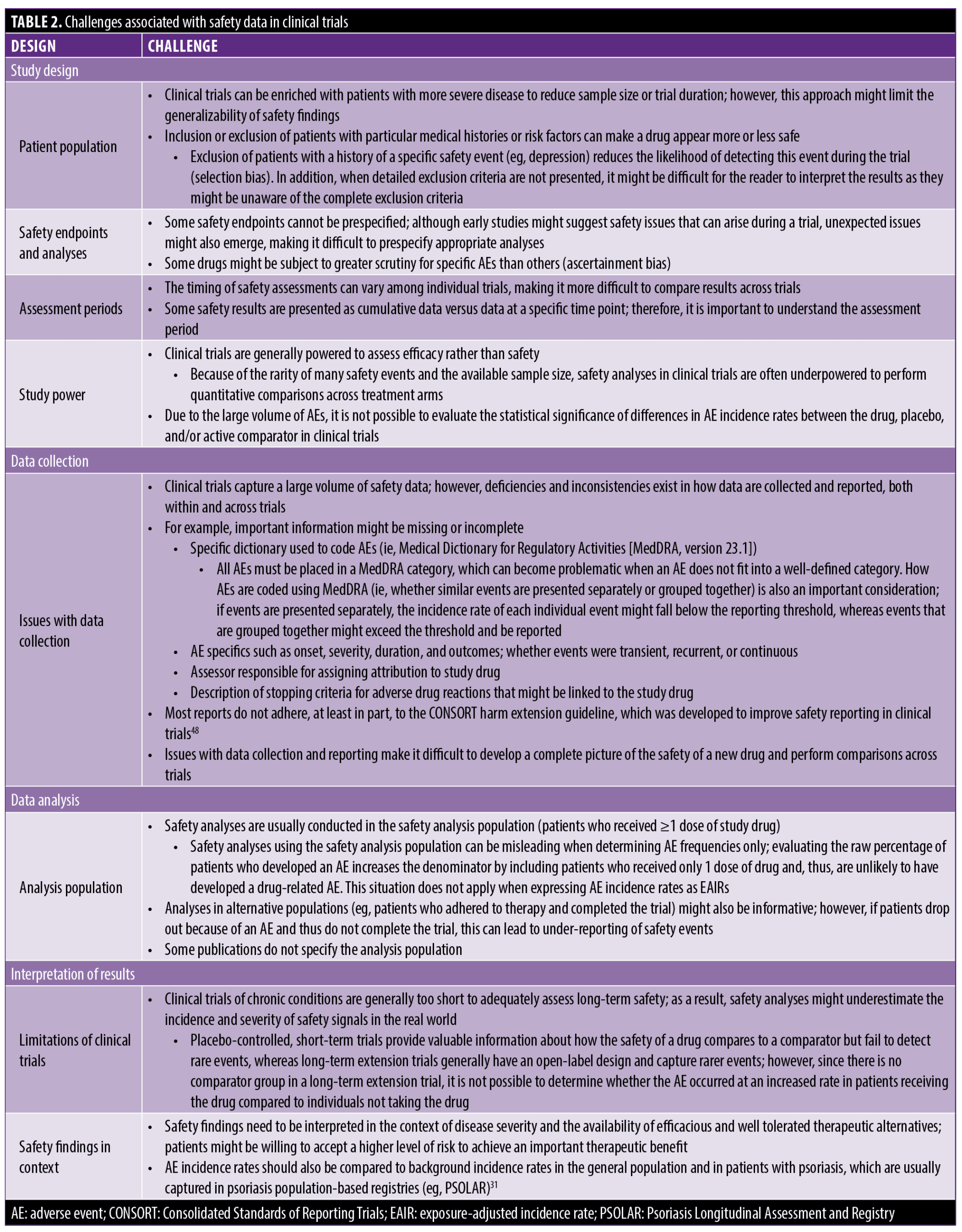
Clinical trials capture a large volume of safety data. However, inconsistencies exist across trials in terms of how safety event data are captured, such as differences in the AE coding approach using the Medical Dictionary for Regulatory Activities (separately vs. grouped) and the specific parameters recorded in relation to each safety event.29 Although safety analyses are generally performed in the safety analysis population (patients who received ≥1 dose of study drug), some publications do not specify the analysis population used.29 These issues with data collection and analysis make it difficult to perform comparisons across trials and develop a complete picture of the safety of a new drug.
Finally, it is important to understand the limitations of clinical trials when interpreting safety data. Randomized controlled trials facilitate comparison of the safety of a new drug versus placebo or an active comparator but can fail to identify rarer events because of the limited sample size and study duration.30 Conversely, a small sample size might inflate incidence rates of some events. In contrast, long-term extension trials usually have an open-label design and lack a comparator, thereby increasing the likelihood of identifying rarer events and better estimating the incidence and severity of safety events in real-world populations.30 Incidence rates of safety events should be compared to background rates in the general population and to rates in patients with psoriasis captured in psoriasis population-based registries (eg, Psoriasis Longitudinal Assessment and Registry [PSOLAR]).31
A 2019 review of published clinical trials illustrates some of the challenges associated with the collection, analysis, and reporting of safety data as described above.29 Among 184 clinical trials in various therapeutic areas published in four high-tier journals, 96 percent reported safety data in the main text. The analysis population tended to vary among trials: 41 percent analyzed AEs in patients receiving one or more dose of study drug, 29 percent in all randomized patients, and 9 percent did not specify the analysis population. Nearly 80 percent of publications reported the number of patients who withdrew from the trial; however, only 35 percent reported whether withdrawals were due to AEs and only 24 percent of this subgroup specified the actual events that resulted in withdrawals. Most safety results were presented for patients with at least one event, with 84 percent of reports providing no information about the number of events that had occurred. In addition, 41 percent of publications reported AE severity, 5 percent reported the duration of at least one AE, and 28 percent included information about the timing of AEs. In 24 percent of reports, it was impossible to discern whether SAEs had not occurred or whether SAEs had occurred but were omitted from the report. A subset of AEs was described in greater detail in 89 percent of trials, but it was neither clear nor consistent how AEs were selected for inclusion in these subsets. Most trials summarized the presence or absence of AEs using frequencies (94%) and percentages (87%); 47 percent reported p-values for these binary outcomes despite the trial not being sufficiently powered to assess the statistical differences. Only 12 percent of reports used graphs to illustrate AE data. None of the included reports referenced the CONSORT extension to harms.32 Based on these findings, the authors concluded that the collection, analysis, and reporting of safety data in clinical trials is inconsistent and suboptimal, with valuable information being either ignored or presented inappropriately.29
Several recent publications have described the safety of approved oral small-molecule and biologic agents in Phase 3 trials of patients with plaque psoriasis.33–39 However, at this time, a comprehensive review or meta-analysis of safety data specific to psoriasis trials has not been identified. Figure 3 depicts three hypothetical psoriasis trials and illustrates the variability that might occur in the reporting of safety data in publications related to these trials. Differences might exist in study design, study duration, type of safety data captured and how incidence rates are expressed (frequencies vs. EAIRs), and whether incidence rates are compared with those reported in patients with psoriasis not receiving the study drug. This variability would make it challenging to determine whether genuine safety differences exist among agents and to compare results across trials.

Adverse events (AEs, treatment-related AEs, SAEs, discontinuations due to AEs, deaths) are usually analyzed in psoriasis trials using descriptive statistics. Adverse events are expressed as crude incidence rates (frequencies and percentages) and exposure-adjusted incidence rates (EAIRs) per 100 person-years (PYs) to account for variable durations of treatment exposure or treatment switching (eg, patients crossing over from placebo to active treatment at designated milestones or vice versa as part of the study design).9,10
Exposure-adjusted incidence rate is calculated as 100 × (number of patients with AE) / (total exposure time for all patients at risk [time to initial AE occurrence for patients with AE + total exposure time for patients without AE]).
As an example, 300 patients each take a drug for two years; three patients experience AEs, one after six months, one after one year, and one after 1.5 years.
Total exposure = (297 patients without AE × two years of treatment) + (one patient with AE × 0.5 year of treatment prior to AE) + (one patient with AE × one year of treatment prior to AE) + (one patient with AE × 1.5 years of treatment prior to AE) = 596 PY; 100 × 3 /596 = EAIR of 0.5 per 100 PY.
Less commonly, clinical trials might express EAIRs per 1,000 PY or 10,000 PY rather than per 100 PY; therefore, it is important to note the denominator when comparing EAIRs across trials. Because exposure is determined based on the time to initial occurrence of an AE, this may have implications when the same AE recurs multiple times because the extended duration of exposure is not considered.
Exposure-adjusted incidence rates should be presented for each AE observed with a new drug, and rates should be compared to those reported in the psoriasis population not receiving the study drug. Exposure-adjusted incidence rates in patients with psoriasis might be captured in patient registries such as PSOLAR,31 and EAIRs also might be compared to placebo (placebo-controlled trials) and/or to approved treatment(s) (active comparator–controlled trials).9,10,33 Psoriasis population-based baseline rates of depression and anxiety, lymphoma, non-alcoholic fatty liver disease, and MACE should be considered.40 Comparison of malignancy incidence rates to those reported in the Surveillance, Epidemiology, and End Results (SEER) program of cancer statistics might also be informative.41
Select AEs of special interest (AESIs) are predefined at the beginning of a clinical trial and are typically related to known risks associated with a specific agent or the class of drugs to which it belongs. Examples of AESIs in patients receiving oral small-molecule or biologic agents for psoriasis might include infections, malignancies, and MACE.5,42 Determination of overall infection rates, as well as the rates of specific infections such as herpes zoster events, is particularly important in understanding the potential for immunosuppression. It is also important to determine whether there are any deaths and to ascertain the details of any deaths that did occur, especially when clinical trials could have been conducted during atypical events such as the global COVID-19 pandemic. In general, AESIs are analyzed in greater detail than other AEs in terms of onset, severity, duration, and outcome, as well as the presence of comorbid conditions and use of prior and/or concomitant medications.5,17
Safety analyses might be conducted in specific patient populations with psoriasis (eg, safety analyses based on age, gender, race, presence of comorbid conditions, and/or use of concomitant medications) to identify safety issues that might be unique to or occur at higher incidence rates in these groups, or when strict eligibility criteria might introduce bias by including only patients who are healthy, young, and do not have obesity.4 Clinical trial results might also be pooled to increase the sample size and identify rarer events.33 A meta-analysis preceded by a systematic review process where rigorous assessment of the validity and heterogeneity of relevant trials are performed might be helpful in building a more complete safety profile rather than relying solely on single trials in isolation.43,44
Adverse events are generally presented in tabular rather than in graphical form, which makes it more challenging to analyze a large volume of data and identify any trends or patterns that might denote safety issues of concern.29 Readers should ensure that there is no selective presentation of AE data in the text versus the tables and figures and, if they have access to the source documentation, ensure that all AEs above the specified cutoff are reported. Ideally, publications should present AEs occurring in at least 1 percent of patients (or the corresponding EAIR/100 PY) in any treatment group, although the practicality of this approach might be influenced by the volume of AEs reported during the trial and any known AEs for the class of medication.
Laboratory parameters in patients with psoriasis should be monitored in accordance with expert-written therapeutic area guidelines, such as the American Academy of Dermatology and National Psoriasis Foundation guidelines and the EuroGuiDerm Guideline and Consensus Statement Development Manual.45–47 Psoriasis trials typically analyze laboratory parameters related to a specific drug class using descriptive statistics. Parameters evaluated include mean change from baseline in select laboratory parameters over time, number of patients experiencing a laboratory abnormality at or above a particular severity level (eg, liver function tests ≥3 times the upper limit of normal [ULN]), shifts from baseline in severity grade of laboratory parameter abnormalities, and discontinuations due to laboratory parameter abnormalities. Laboratory parameter results for the new drug are usually compared to placebo and/or approved treatment(s)9,10,33 and are also typically presented in relation to normal reference ranges. This can result in some parameters, such as lipids and liver enzymes, exceeding the ULN because baseline levels of these parameters are known to be higher in patients with psoriasis. Similar to AEs, laboratory parameters might be analyzed in pooled clinical trials to better understand any abnormalities observed in individual trials.33 Laboratory parameters are typically presented in graphical and tabular form.
Conclusion
Clinical trials provide important safety information about new and established drugs used to treat psoriasis. However, numerous challenges exist in relation to the collection, analysis, reporting, and interpretation of safety data. Clinical trials are generally limited in size and duration, exclude high-risk patient populations, and have limited statistical power to detect rare but potentially serious AEs, thereby limiting the generalizability of safety findings from clinical trials to real-world situations.30 A thorough understanding of the interpretation of safety data, especially the appropriate use of EAIRs in relation to AEs, as well as the importance of comparing incidence rates to those reported in the general population and in patients with psoriasis, is vital to making a well-informed assessment of the safety of a new drug. This knowledge is also helpful in understanding the limitations inherent when making side-by-side comparisons of safety data from different clinical trials.
References
- Umscheid CA, Margolis DJ, Grossman CE. Key concepts of clinical trials: a narrative review. Postgrad Med. 2011;123(5):194–204.
- Phillips R, Sauzet O, Cornelius V. Statistical methods for the analysis of adverse event data in randomised controlled trials: a scoping review and taxonomy. BMC Med Res Methodol. 2020;20(1):288.
- DiRuggiero D, Trickett C, Hippeli L, et al. Review of statistical considerations and data imputation methodologies in psoriasis clinical trials. J Clin Aesthet Dermatol. 2024;17(7-8 suppl 1):S15–S24.
- Zink RC, Marchenko O, Sanchez-Kam M, et al. Sources of safety data and statistical strategies for design and analysis: clinical trials. Ther Innov Regul Sci. 2018;52(2):141–158.
- Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–243.
- Common Terminology Criteria for Adverse Events (CTCAE) v5.0. 2017. Available at: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5x7.pdf. Accessed January 2, 2024.
- Nebeker JR, Barach P, Samore MH. Clarifying adverse drug events: a clinician’s guide to terminology, documentation, and reporting. Ann Intern Med. 2004;140(10):795–801.
- NIA Adverse Event and Serious Adverse Event Guidelines. 2018. Available at: https://www.nia.nih.gov/sites/default/files/2018-09/nia-ae-and-sae-guidelines-2018.pdf. Accessed May 23, 2024.
- Armstrong AW, Gooderham M, Warren RB, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, placebo-controlled Phase 3 POETYK PSO-1 trial. J Am Acad Dermatol. 2023;88(1):29–39.
- Strober B, Thaçi D, Sofen H, et al. Deucravacitinib versus placebo and apremilast in moderate to severe plaque psoriasis: efficacy and safety results from the 52-week, randomized, double-blinded, Program fOr Evaluation of TYK2 inhibitor psoriasis second Phase 3 trial. J Am Acad Dermatol. 2023;88(1):40–51.
- Herson J. Data and Safety Monitoring Committees in Clinical Trials. Boca Raton, FL: Taylor & Francis; 2017.
- Yao B, Zhu L, Jiang Q, et al. Safety monitoring in clinical trials. Pharmaceutics. 2013;5(1):94–106.
- O’Connor NR. FDA boxed warnings: how to prescribe drugs safely. Am Fam Physician. 2010;81(3):298–303.
- Xeljanz [package insert]. New York, NY: Pfizer Inc.; 2021.
- Olumiant [package insert]. Indianapolis, IN: Lilly USA, LLC; 2018.
- Rinvoq [package insert]. North Chicago, IL, USA: AbbVie Inc.; 2024.
- Gadina M, Chisolm DA, Philips RL, et al. Translating JAKs to Jakinibs. J Immunol. 2020;204(8):2011–2020.
- Skyrizi [package insert]. North Chicago, IL, USA: AbbVie Inc.; 2024.
- Ilumya [package insert]. Cranbury, NJ: Sun Pharmaceutical Industries, Inc.; 2022.
- Tremfya [package insert]. Horsham, PA: Janssen Biotech, Inc.; 2023.
- Humira [package insert]. North Chicago, IL: AbbVie; 2023.
- Enbrel [package insert]. Thousand Oaks, CA: Immunex Corporation; 2023.
- Infliximab [package insert]. Horsham, PA: Janssen Biotech, Inc.; 2021.
- Cosentyx [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2023.
- Taltz [package insert]. Indianapolis, IN: Eli Lilly and Company; 2024.
- Siliq [package insert]. Bridgewater, NJ: Bausch Health US LLC; 2020.
- Bimzelx [package insert]. Smyrna, GA: UCB, Inc.; 2023.
- Stelara [package insert]. Horsham, PA: Janssen Biotech, Inc; 2024.
- Phillips R, Hazell L, Sauzet O, et al. Analysis and reporting of adverse events in randomised controlled trials: a review. BMJ Open. 2019;9(2):e024537.
- Singh S, Loke YK. Drug safety assessment in clinical trials: methodological challenges and opportunities. Trials. 2012;13:138.
- Bissonnette R, Gottlieb AB, Langley RG, et al. Signal detection and methodological limitations in a real-world registry: learnings from the evaluation of long-term safety analyses in PSOLAR. Drug Saf. 2021;44(6):699-709.
- Junqueira DR, Zorzela L, Golder S, et al. CONSORT Harms 2022 statement, explanation, and elaboration: updated guideline for the reporting of harms in randomised trials. BMJ. 2023;381:e073725.
- Strober B, Blauvelt A, Warren RB, et al. Deucravacitinib in moderate to severe plaque psoriasis: pooled safety and tolerability over 52 weeks from two Phase 3 trials (POETYK PSO-1 and PSO-2). J Eur Acad Dermatol Venereol. 2024;38(8):1543–1554.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: Results of a Phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73(1):37–49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a Phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173(6):1387–1399.
- Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled Phase 3 trials. Lancet. 2018;392(10148):650–661.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, Phase 3 trials. Lancet. 2017;390(10091):276–288.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the Phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–417.
- Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: Results from the Phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76(3):418–431.
- Greb JE, Goldminz AM, Elder JT, et al. Psoriasis. Nat Rev Dis Primers. 2016;2:16082.
- Surveillance, Epidemiology, and End Results (SEER) Program. 2024. Available at: https://seer.cancer.gov/. Accessed August 29, 2024.
- Rungapiromnan W, Yiu ZZN, Warren RB, et al. Impact of biologic therapies on risk of major adverse cardiovascular events in patients with psoriasis: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. 2017;176(4):890–901.
- Nast A, Jacobs A, Rosumeck S, et al. Efficacy and safety of systemic long-term treatments for moderate-to-severe psoriasis: a systematic review and meta-analysis. J Invest Dermatol. 2015;135(11):2641–2648.
- Westergren T, Narum S, Klemp M. Biases in reporting of adverse effects in clinical trials, and potential impact on safety assessments in systematic reviews and therapy guidelines. Basic Clin Pharmacol Toxicol. 2022;131(6):465–473.
- Menter A, Gelfand JM, Connor C, et al. Joint American Academy of Dermatology-National Psoriasis Foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82(6):1445–1486.
- Nast A, Smith C, Spuls PI, et al. EuroGuiDerm Guideline on the systemic treatment of Psoriasis vulgaris—part 1: treatment and monitoring recommendations. J Eur Acad Dermatol Venereol. 2020;34(11):2461–2498.
- Florek A, Armstrong AW. Relevant laboratory tests and therapeutic monitoring in psoriasis. In: Adebajo A, Boehncke W-H, Gladman DD, Mease PJ, eds. Psoriatic Arthritis and Psoriasis: Pathology and Clinical Aspects. Switzerland: Springer International Publishing; 2016:221–226.
- Moher D, Hopewell S, Schulz KF, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c869.