Resident’s Forum
Section Editor: Jerry Tan, MD
Resident Section Editors: Sanjay Bhambri, DO, and Joshua Zeichner, MD
Anne Han, MD; Joshua A. Zeichner, MD
Dr. Han is Clinical Dermatopharmacology Fellow, Department of Dermatology, Mt. Sinai Medical Center, New York, New York. Dr. Zeichner is Chief Resident, Department of Dermatology, Mt. Sinai Medical Center, New York, New York. Section Editor: Jerry Tan, MD, FRCPC, is Adjunct Professor, University of Western Ontario, London, Ontario; President, Windsor Clinical Research Inc., Windsor, Ontario; and Consultant, Windsor Regional Hospital, Windsor, Ontario, Canada. He is also in private practice.
Abstract
The bullous diseases comprise a heterogeneous group of skin disorders with distinct clinical and histological findings. They are characterized histologically by clefts at varying depths in the skin and are pathologically caused either by congenital defects or autoantibodies. Autoimmune bullous disorders are chronic conditions with significant morbidity and mortality in untreated patients. With the advent of immunosuppressive medications, mortality from these diseases has decreased significantly. However, complications from therapy itself are common causes of morbidity in these patients. Therefore, treatment of autoimmune bullous diseases is a challenge, as patients must remain on chronic medications with side effects that limit their use. This article aims to provide a practical approach to understanding the available medications for the treatment of autoimmune bullous diseases.
Armamentarium of Medications
The current treatment armamentarium includes topical and systemic glucocorticoids, cytotoxic agents, and other therapies, such as dapsone, intravenous immunoglobulin (IVIG), and rituximab (Table 1). Use of these drugs for bullous diseases is largely off label from their approved indications. In addition, much of the data available is based on small series, prospective studies, and case reports, as the rarity and severity of these conditions preclude the development of large, double-blind, clinical trials. Interpretation of the available data, knowledge of the pathophysiology of the diseases and pharmacology of the drugs, and experience in managing potential complications of these drugs are important components in the successful treatment of autoimmune bullous disorders.
{kind=link}
Glucocorticoids. Glucocorticoids are the mainstay of treatment for most bullous disorders. They have anti-inflammatory and immunosuppressive effects from the inhibition of the production of proinflammatory cytokines. They diminish the number of circulating T-cell lymphocytes and reduce their responsiveness to antigens. In addition, glucocorticoids decrease antibody production.[1]
Adverse effects from glucocorticoids arise both from their long-term use and abrupt discontinuation. Immunosuppression increases patients’ risks of developing infections. In addition, prolonged use may lead to osteoporosis, osteonecrosis, cushingoid fat redistribution, and acid reflux disease. Patients may also develop electrolyte imbalances, hyperglycemia, hypertriglyceridemia, proximal myopathy, hyperactivity, glaucoma, and cataracts.[1] Physicians prescribing patients long-term glucocorticoid therapy should order baseline and annual bone densitometry tests and recommend calcium and vitamin D supplements. Bisphosphonates may be added in patients with pre-existing osteopenia or osteoporosis. Blood pressure should be monitored regularly as well as blood glucose, triglycerides, electrolytes, and tuberculin skin testing.[2]
The most significant adverse event associated with acute withdrawal of corticosteroids is hypothalamic-pituitary-adrenal (HPA) axis suppression. Exogenous steroid use provides a negative feedback loop that inhibits endogenous steroid production. The risk of HPA axis suppression can be minimized with single morning doses that parallel the body’s circadian cortisol production. In addition, alternate-day dosage when tapering the drug can reduce systemic complications.[1]
Azathioprine. Azathioprine is a purine analog used as a steroid-sparing agent for autoimmune bullous diseases. It is a prodrug, which is converted first to 6-mercaptopurine, then to 6-thioguanine, the active metabolite. Azathioprine acts during the S-phase of the cell cycle and inhibits the formation of adenine and guanine nucleotides. Dosages range from 3 to 4mg/kg. Onset of action is slow, as it usually takes between 6 to 8 weeks to take effect. Initial monitoring should consist of complete blood count (CBC) and liver enzymes every two weeks.[3]
Side effects from azathioprine are uncommon, but include gastrointestinal toxicity, hepatotoxicity, alopecia, and pancreatitis. In addition, lymphoproliferative diseases and infection rates may be elevated.[1] Azathioprine is metabolized by several enzymes, including hypoxanthine guanine phosphoribosyltransferase (HGPRT), xanthine oxidase (XO), and thiopurine methyltransferase (TPMT). Since genetic polymorphisms exist, prescribers must measure TPMT levels prior to therapy. In those with low levels of TPMT, the drug will be more readily metabolized by the HGPRT pathway, leading to higher drug levels. Therefore, appropriate dosing depends on this enzyme level. Another consideration when prescribing azathioprine is to avoid certain drugs, such as allopurinol, which inhibits XO and shunts azathioprine metabolism toward the HGPRT pathway. This increases the risk of bone marrow suppression.[3,4]
Mycophenolate mofetil. Myco-phenolate mofetil (MMF) inhibits de-novo purine synthesis by noncompetitively inhibiting inosine monophosphate dehydrogenase (IMPDH). Both B-cell and T-cell lymphocytes are most affected by MMF because these cells lack a purine salvage pathway.1 Since the salvage pathway is still, this drug may be considered safer than other immunosuppressive agents. Dosages range from 0.5 to 1.5g twice daily. After ingestion, MMF is converted to mycophenolic acid, which is metabolized by the liver.
MMF is well tolerated, with the most common side effect being gastro-intestinal distress. Patients may experience a dose-related and reversible anemia, leukopenia, and thrombo-cytopenia. Additionally, there is a slight increase in the incidence of infections. Physicians must monitor CBC and liver enzymes monthly.[1]
Cyclophosphamide. Cyclo-phosphamide is an alkylating agent that binds to DNA nonspecifically during the cell cycle. This nitrogen mustard derivative arrests the cell cycle and induces apoptosis in cells with a high mitotic rate, such as lymphocytes. It is metabolized by the hepatic cytochrome P450 system. Laboratory monitoring consists of renal function, CBC with platelets, and urinalysis.[5]
The toxicity of cyclophosphamide is significantly higher than that of azathioprine and MMF, although many patients do not experience serious side effects. Acute myelosuppression is common. Other side effects include mucosal ulcers, alopecia, nephrotoxicity, cardiotoxicity, hepatotoxicity, and interstitial lung fibrosis. Rarely, male patients may develop azoospermia.[1]
Hemorrhagic cystitis occurs in up to 40 percent of patients and is associated with development of transitional cell carcinoma. This toxicity is due to the acrolein metabolite of cyclophos-phamide. The risk of cystitis may be reduced with high fluid intake, frequent urination, mesna, and monitoring for hematuria. The drug must be discontinued if red blood cells are found in the urine.[1]
Methotrexate. Methotrexate is an antimetabolite that suppresses DNA and RNA synthesis during the S-phase of cell cycle. The drug competitively inhibits dihydrofolate reductase, which decreases the formation of tetrahydrofolate, a cofactor important in de-novo purine and thymidylate synthesis. Patients should be started on a low dose (often with an initial test dose) and cautiously increased every week to the target dose. Lab monitoring consists of CBCs, electrolytes, and renal- and liver-function tests. Additionally, methotrexate is a teratogen, affecting both egg and sperm production.[1]
The most common adverse effect of methotrexate is hepatotoxicity. Patients who have a cumulative dose >1.5g should undergo liver biopsy.[6] Additionally, the drug may cause bone marrow suppression, ulcers, alopecia, interstitial pneumonitis and fibrosis, and nephrotoxicity. Predisposing factors for toxicity include alcohol consumption, renal insufficiency, diabetes, and increasing cumulative doses. Further, patients should avoid drugs that inhibit folic acid metabolism. Certain drugs may also displace plasma proteins and increase methotrexate levels. These include tetracyclines, phenytoin, phenothiazines, chloramphenicol, nonsteroidal anti-inflammatory drugs, and salicylates.[1] An acute methotrexate overdose may cause bone marrow suppression or gastrointestinal mucositis. In these cases, folinic acid can be administered as a rescue treatment.
Cyclosporine. Cyclosporine is an immunosuppressive agent with anti-T-cell lymphocyte activity. It forms a complex with cytoplasmic cyclophilin and blocks its ability to activate calcineurin. Activated calcineurin normally phosphorylates NFAT-1, a transcription factor that initiates IL-2 production and promotes proliferation of helper and cytotoxic T-cells.[7] Laboratory monitoring consists of renal- and liver-function tests, CBCs, electrolytes, lipid profile, and blood pressure.[1]
Cyclosporine has many potential side effects. The most common problems are electrolyte abnormalities, such as hyperkalemia, hyperuricemia, and hypomagnesemia. The drug is metabolized by the hepatic cytochrome P450 system and has the potential for many drug interactions. Cyclosporine is nephrotoxic, which is dose related and may initially be reversible. However, long-term use often leads to irreversible kidney damage and hypertension. Renal toxicity may be potentiated by concomitant use of aminoglycosides, nonsteroidal anti-inflammatory drugs, amphotericin B, and vancomycin. Other side effects include tremors, hirsutism, hyperlipidemia, hypertension and gingival hyperplasia.[8]
Dapsone. Dapsone is a sulfone antibiotic with anti-inflammatory activity primarily against polymorphonuclear leukocytes. The drug inhibits neutrophil toxicity and chemotaxis by blocking myeloperoxidase activity.
Dapsone commonly causes a hemolytic anemia and methemoglobulinemia in varying degrees in all patients. It is dose-related, but is most severe in patients with glucose-6-phosphate dehydrogenase deficiency.[1] These patients are extremely sensitive to oxidative stress from dapsone metabolites. Cimetidine and vitamin E may provide some protection against methemoglobinemia. Dapsone is also associated with an idiosyncratic peripheral motor neuropathy and psychosis. Agranulocytosis is a rare, but serious reaction occurring in the first three months of therapy.[9,10] Finally, a dapsone hypersensitivity syndrome may develop, comprising severe mononucleosis-like reactions, such as fever, erythroderma, hepatitis, eosinophilia, or death.[11] CBCs should be monitored weekly for the first month, monthly for six months, and semiannually thereafter. Renal- and liver-function tests should be obtained every three months for potential toxicity.[1]
Tetracycline and niacinamide. Tetracycline and niacinamide may be combined to treat autoimmune blistering disorders, such as bullous pemphigoid. Tetracycline is an antibiotic that works through inhibition of the 30S ribosomal subunit. In addition to antibiotic activity, it possesses anti-inflammatory properties. Niacinamide is the amide of niacin (vitamin B3) and also is anti-inflammatory. The mechanism of action of their combined, immune-modulating properties is unclear. However, it is known that they inhibit neutrophil and eosinophil chemotaxis, which may downgrade the humoral response. The drugs may be dosed between 1 to 2g/day of tetracycline and 1 to 2g/day of niacinamide.[12]
Side effects from the drugs include gastrointestinal distress, pseudotumor cerebri, and photosensitivity from tetracycline. In addition, permanent discoloration may develop during tooth development, so tetracycline should not be given to children under nine years of age. Niacinamide has been associated with pruritus and flushing, especially if consumed with alcohol. Patients also may report nausea and headache and there may be drug-drug interactions, especially with lipid-lowering agents.[12]
Intravenous immunoglobulin (IVIG). High-dose IVIG is a purified, human source of immunoglobulin G (IgG) from pooled plasma. Preparation of IVIG undergoes three independent processes for the inactivation and removal of viral contaminants, such as hepatitis and human immunodeficiency virus. It is administrated as a slow, IV infusion at 2g/kg per cycle divided over 3 to 5 consecutive days. Treatment is repeated every four weeks. The mechanism is unknown although several theories exist. These include inhibition of Fc receptors, inhibition of autoantibody formation and neutralization of autoantibodies, complement and cytokine inhibition, and inhibition of the Fas-FasL interaction and apoptosis, and enhanced steroid sensitivity.[13,14] The combination of IVIG with traditional immuno-suppressive agents may decrease antibody levels and improve clinical efficacy and offset its rebound following antibody depletion by IVIG.[15]
There are several potential complications from IVIG. Patients with renal insufficiency are at risk for renal failure, so infusions must be given slowly and cautiously. In addition, transfusion-related acute lung injury (TRALI) is rare and if there is a previous history of infusion reactions, premedication with steroids or antihistamines is indicated. Other adverse effects are rare and include fever, headache, myalgia, nausea, tachycardia, hemolysis, aseptic meningitis, thrombotic event, and anaphylaxis in patients with IgA deficiency due to trace amounts of IgA in the preparation. Serum levels of IgA and hepatitis panel are screened before initiation of therapy. CBC and complete metabolic panel, including liver function tests, are monitored prior to each cycle. IVIG is very expensive and generally reserved for patients who do not respond or have serious adverse events from standard therapies.[14]
Rituximab. Rituximab is a genetically engineered, chimeric, murine/human monoclonal antibody directed against CD20 antigen on the cell surface of normal and malignant B-cell lymphocytes. It is administered as an IV infusion, and premedication with acetaminophen and diphenhydramine should be considered to avoid infusion reactions. Several theories exist on the drug’s mechanism of action. Among them is the drug’s ability to inhibit autoantibody-producing B-cells in the immune system.[16]
Rituximab has several black box warnings due to serious adverse events that had fatal outcomes. These fatalities resulted from infusion reactions usually occuring within the first 24 hours of the first infusion, acute renal failure from tumor lysis syndrome, severe mucocutaneous reactions, and progressive multifocal leukoencephalopathy due to JC virus infection.
Management of Pemphigus Vulgaris
Pemphigus vulgaris is a serious bullous autoimmune disease that causes acantholysis of the skin and mucous membranes and often leads to death if not treated appropriately. The disease occurs more commonly in Jewish people and those of Mediterranean descent. Pemphigus vulgaris usually starts as painful oral lesions before progressing to a generalized skin eruption of flaccid bullae on normal skin. Since blisters rupture easily, only painful erosions are seen in some patients. Direct immunofluorescence staining reveals IgG and C3 deposits in the intercellular substance of the epidermis in lesional and perilesional skin. Titers of IgG, the autoantibody against desmoglein 3, usually correlate with disease activity. Thus, clinical response can be monitored by antibody titers (antibodies have a half life of about three weeks). This disease is known for its remitting/relapsing course, and lifelong therapy is often required.
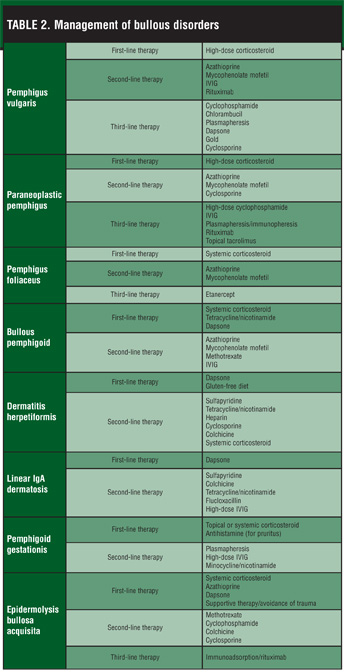
The primary goal of treatment in pemphigus vulgaris is reducing the synthesis of autoantibodies by the immune system. Systemic glucocorticoids are the first-line treatment of this disease in reducing the synthesis of pathogenic antibodies (Table 2). Its advent in the 1950s revolutionized the prognosis of this disease, improving mortality rates from 90 to 10 percent. The medication works relatively quickly and safely. Patients should be started on 1mg/kg daily. A good clinical response is usually seen in 2 to 3 months, after which the drug can be reduced to 40mg daily and subsequently tapered over 6 to 9 months to a maintenance dose of 5mg every other day. Tapering is achieved either by reducing the dose by 10mg per month initially and then by 5mg per month or by an alternating two-day dose regimen of 40/20, 40/0, 30/0, 20/0, 15/0, 10/0, and 5/0mg followed by 5/0mg for maintenance.17 Pulse steroid therapy involves IV methylprednisolone given 1g/day for three consecutive days. It allows a rapid response to occur without the long-term side effects of chronic steroid use. A retrospective review of patients with severe, refractory, pemphigus vulgaris showed long-lasting benefits of pulse steroid therapy.[18]
{kind=link}
The addition of a second-line therapy is indicated to improve efficacy, reduce side effects of steroid use, and treat relapse during prednisone tapering. The addition of a second-line therapy is also indicated if the disease flares. Second-line therapies consist of the antimetabolites-azathioprine and MMF-as well as IVIG and rituximab. The antimetabolites are prescribed at full doses for 2 to 3 years to induce a durable remission. Azathioprine is more commonly used than MMF because it has been around longer and is more affordable. However, recent studies demonstrate that combination therapy with MMF and glucocorticoids is more effective and safer than with azathioprine as an adjuvant to steroid therapy.[19] MMF has also been shown to be an effective monotherapy in two case reports.[20,21] IVIG has emerged as a rapid, safe, and effective, though very expensive, adjuvant treatment in patients who are refractory to or have contraindications to conventional treatment. IVIG may also play a role in acute control when plasmapheresis is not indicated.[22] Serum studies show that IVIG can selectively and markedly reduce abnormal antibody levels without decreasing the levels of normal antibodies.[23] Rituximab has been shown to induce a prolonged clinical remission after a single course of four weekly treatments in the largest review of 12 patients.[16] Several reports of serious infections resulting from pemphigus patients treated with rituximab exist.[24] Rituximab is a promising therapeutic option but only for selected patients with refractory or life-threatening disease due to its high cost, serious and potentially fatal complications, and limited knowledge of long-term adverse events.
The most effective agent for inducing remission by inhibiting antibody synthesis is cyclosphosphamide.[25] However, this drug is used as a third-line therapy due to its high side effect profile. Chlorambucil can be prescribed as a substitute for cyclophosphamide in the event the patient develops hemorrhagic cystitis from the latter drug.[2] Plasmapheresis is used to rapidly reduce antibody levels in patients with extensive or accelerated disease. It must be combined with systemic corticosteroids and cyclophosphamide to prevent a rebound flare of the disease.22 Dapsone has been shown to reduce corticosteroid dependency in the maintenance phase of pemphigus.[26] In a retrospective review, gold was effective in 62 percent of patients but its use has waned in recent years due to concerns regarding toxicity and inferior efficacy compared to newer treatments.[27] However, gold may have a use in patients of reproductive age, as it lacks carcinogenic and infertility effects that most other immunosuppressive drugs exhibit. There is good evidence that cyclosporine does not have any advantage as adjuvant therapy and should not be used, as complications are common from this drug. Temporary relief of pain or inflammation by topical steroids has little effect on the course of the disease.
Management of Paraneoplastic Pemphigus
Paraneoplastic pemphigus (PNP) is an acquired, mucocutaneous disease associated with an underlying neoplasm, the most common being non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, and Castleman’s disease. Starting at a mean age of 60, the disease is characterized by painful, progressive stomatitis and may also include a highly variable cutaneous eruption of diffuse erythema, vesiculobullous lesions, papules, plaques, erosions or ulcerations. PNP is often fatal, approaching rates of 90 percent, especially when associated with a malignancy. Causes of death often result from sepsis as well as respiratory failure, due to the direct effects of the disease on skin integrity and respiratory epithelium.[28] IgG and complement are deposited on epithelial cell surfaces and along the basement membrane zone (BMZ) leading to acantholysis, dyskeratosis, and vacuolar interface dermatitis. Indirect immunofluorescence reveals binding of pathogenic antibodies to the cell surface of stratified squamous epithelia as well as simple, columnar, and transitional epithelia. Antibodies are directed against a group of molecules belonging to the plakin family, which play an important role in intermediate filament attachment. Target antigens include desmoglein 3, desmoglein 1, envoplakin, periplakin, desmoplakin I, desmoplakin II, and bullous pemphigoid antigen I. The gold standard for diagnosis is immunoprecipitation.[29]
The initial treatment goal of PNP is aimed at treating any superinfection. Otherwise, management involves treating the underlying neoplasm and suppression of the immune system. Surgical resection of benign neoplasms may improve skin disease, but patients with malignant neoplasms may be resistant to treatment. First-line therapy is high-dose corticosteroids,[30] followed by steroid-sparing agents, such as azathioprine, MMF, and cyclosporine (Table 2). Other therapeutic options include high-dose cyclophosphamide without stem cell rescue, IVIG, plasmapheresis, and immunopheresis. Rituximab has been effective in a case of PNP associated with CD20-positive follicular lymphoma.[31] Topical tacrolimus was used successfully in a patient with refractory oral stomatitis.[32] In general, polymorphic skin lesions are more responsive than mucosal lesions, although overall response to treatment is often poor.
Management of Pemphigus Foliaceus
Pemphigus foliaceus (PF) is characterized by superficial scaling erosions appearing on the face and scalp and later involving the chest and back, without mucous membrane involvement. It is more prevalent in Africa, and endemic forms have also been described, such as fogo selvagem occurring in Brazil. Immunopathologic studies reveal subcorneal acantholysis and tissue-bound and circulating antidesmoglein 1 antibodies. PF may be drug induced or drug triggered. The drugs most commonly implicated are penicillamine, captopril, piroxicam, penicillin, and topical imiquimod.[33] Spontaneous remission is common.
The diagnosis and treatment of PF are similar to PV, but PF is generally milder and requires less aggressive treatment. While the first line of treatment is systemic corticosteroids, not every patient requires immediate treatment, as some patients have very limited disease and can therefore benefit from topical corticosteroids alone.[22] Second-line treatments include MMF[34] and azathioprine (Table 2). A recent case study demonstrated that etanercept is also an effective steroid-sparing agent.[35]
Management of Bullous Pemphigoid
Bullous pemphigoid is the most common autoimmune bullous disease, usually presenting in patients over the age of 60 as a self-limiting condition. It begins with a prodrome of erythematous papular or urticarial-type lesions. After several weeks to months, these lesions evolve into a generalized eruption of large, tense, firm-topped bullae that do not rupture easily. Eventually, they collapse and crust over. The only symptom is moderate-to-severe pruritus. Later on, the patient may experience tenderness of the eroded lesions. On histopathology, bullous pemphigoid appears as subepidermal blisters. Immunopathology reveals linear IgG and C3 deposits along the BMZ. Circulating antibasement membrane IgG antibodies for two types of antigens, BP1 (230kd) and BP2 (180kd), are detected by indirect immunofluorescence in 70 percent of patients. Mortality of BP patients is more likely related to advanced age and associated medical conditions than to disease-specific factors.[22]
When treating bullous pemphigoid, considering patient age is important, as risks of therapy are heightened in the elderly population. Patients with localized disease can be managed successfully with topical or intralesional corticosteroids (Table 2). For extensive disease, systemic corticosteroid, dosed one-half to three-fourths mg/kg/d, as monotherapy is sufficient to cause remission.2 Higher dosages are rarely needed and osteoporosis prevention should be implemented. Patients who are unable to tolerate or who have contraindications to corticosteroids can be treated with tetracycline alone or in combination with nicotinamide.[36] This treatment is also attractive for younger patients who may need long-term immunosuppressive therapy. Dapsone is another alternative to steroids and is particularly useful when histologic examination reveals a predominance of neutrophils.[22]
Second-line therapy consists of the cytotoxic medications, such as azathioprine, MMF, and methotrexate. While both demonstrate similar efficacy, MMF is the preferred agent when compared to azathioprine because the former has significantly less liver toxicity.[37] Methotrexate is effective in small doses as a sole therapy, and a recent study has shown it may work better than prednisone in patients with generalized BP.[38] High-dose IVIG is considered safe and extremely effective in resistant disease.[39] Patients usually receive 2 to 4 doses total to obtain remission. It can be combined with an immunosuppressant, such as azathioprine or mycophenolate, to offset the antibody rebound effect that is sometimes observed with IVIG treatment.[15]
Management of Dermatitis Herpetiformis
Dermatitis herpetiformis is a chronic, recurrent, intensely pruritic eruption occurring symmetrically on extensor areas of the extremities and trunk. The lesions consist of erythematous papules, wheals, vesicles, and bullae arranged in groups. Scratching results in excoriations, and postinflammatory dyspigmentation occurs after healing. The disease is associated with IgA deposits in the skin and gluten-sensitive enteropathy. Small bowel malabsorption is detected in 10 to 20 percent of patients with dermatitis herpetiformis. The most common age of onset is 30 to 40 years of age, although children may also be at risk. Men are twice as likely to be affected as women.[22]
Dapsone is the only drug approved for use in dermatitis herpetiformis (Table 2).[40] It is started at 50mg daily and the dose is adjusted weekly to maintain adequate control of signs and symptoms. Pruritus improves within the first two days and skin lesions within the first week. A gluten-free diet (GFD) is extremely helpful in the management of this disease. GFD is aimed at the cause rather than the symptoms of the disease. As a result, it can decrease the dosage of dapsone needed, clear lesions leading to permanent remission in a subset of patients as well as improve gastrointestinal symptoms that dapsone cannot.[41] Sulfapyridine is an alternative choice for patients intolerant of dapsone.[42] Nicotinamide, tetracycline, heparin, cyclosporine, colchicine, and systemic corticosteroids have been shown to provide some benefit to this disease.[43–46]
Management of Linear IgA Dermatosis
Linear IgA dermatosis (LAD) is an acquired bullous disease of the skin and mucous membranes that is characterized by linear deposition of IgA along the BMZ. The eruption may be caused by a drug, such as vancomycin, and the eruption appears as arcuate or grouped vesicles and bullae distributed symmetrically on extensor surfaces. Mucosal involvement occurs in 50 percent of cases. The disease is divided into adult linear IgA bullous dermatosis and chronic bullous disease of childhood, which are identical disease processes that present at different ages. Circulating autoantibodies against various epidermal basement membrane antigens have been found.[22]
Dapsone is the preferred drug of treatment for LAD, which responds rapidly within the first few days (Table 2). Since this disease tends to be chronic, long-term therapy with monitoring of potential complications, including hemolysis, methemoglobinemia, and other more serious side effects, is important. Prednisone as an adjuvant will result in clinical improvement and lower the amount of dapsone needed. If patients cannot tolerate dapsone, sulfapyridine may be substituted. Other second-line therapies include colchicine47 and the combination of tetracycline and niacinamide.[48] Tetracycline should not be used in children as it can cause permanent discoloration of teeth. Flucloxacillin has been used as a safer alternative to treat chronic bullous disease of childhood.[49] A recent study showed a partial response to high-dose IVIG in an adult patient with LAD.[50]
Management of Pemphigoid Gestationis
Pemphigoid (herpes) gestationis is a rare, intensely pruritic, polymorphic eruption of vesicles, bullae, and urticarial plaques. It usually occurs during the second or third trimester of pregnancy, and may exacerbate shortly after delivery before resolving several weeks or months later. Pemphigoid gestationis is associated with a high risk of premature birth.[51] It is very similar to BP and occurs when the herpes gestationis factor—the pathogenic antibody against a domain on BPAG2 antigen in the BMZ—causes formation of subepidermal blisters with an inflammatory infiltrate rich in eosinophils.
Because the disease is self limited, treatment is aimed at symptomatic relief of the pruritus and suppression of bullae formation and usually consists of a topical or systemic corticosteroid in combination with an antihistamine (Table 2). Newborns may also develop bullous lesions similar to those of their mother by passive transfer of the antibody across the placenta. These lesions are highly transient and do not require treatment. For cases not responsive to steroids, other options include plasmapheresis,52 high-dose IVIG,[53,54] and as combination therapy with minocycline and nicotinamide.[55] Other drugs currently used in the treatment of BP may be explored, but appropriate consideration of contraindications during pregnancy or breastfeeding is important prior to treatment.
Management of Epidermolysis Bullosa Acquisita
Epidermolysis bullosa acquisita (EBA) is a chronic, subepidermal bullous disease associated with autoimmunity to the noncollagenous (NC1) domain of type VII collagen within the anchoring fibrils in the BMZ. The trauma-prone areas of the skin, such as extensor surfaces of elbows, knees, ankles, and buttocks, are most commonly affected. It primarily involves the skin, but can also affect the mucous membranes. The three presentations of EBA are classic mechanobullous, bullous pemphigoid-like, and cicatricial pemphigoid-like.[22]
Treatment of EBA is difficult, particularly in patients with classic mechanobullous presentation. A commonly used initial regimen is systemic glucocorticoids with either (or both) azathioprine or dapsone as steroid-sparing agents (Table 2).[22] Other treatments include methotrexate and cyclophosphamide, which are somewhat helpful in the inflammatory BP-like form of the disease. Some EBA patients improve on high doses of colchicine[56] and cyclosporin. Supportive therapy and avoidance of physical trauma is warranted in all patients with EBA. More recently, combination treatment with immunoadsorption and rituximab for mechanobullous EBA has shown complete clinical remission and stable disease in some patients.[57]
Conclusion
The management of autoimmune bullous diseases is challenging. With proper understanding of the therapeutic agents as well as close monitoring and treatment of potential complications, dermatologists can confidently prescribe systemic medications. This article equips the clinician with important information to implement a practical approach to the treatment of bullous diseases.
References
1. Wolverton SE. Systemic corticosteroids. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. Philadelphia: WB Saunders;2001:109–138.
2. Mutasim DF. Management of autoimmune bullous diseases: pharmacology and therapeutics. J Am Acad Dermatol. 2004;51(6):859–877.
3. Younger IR, Harris WS, Colver GB. Azathioprine in dermatology. J Am Acad Dermatol. 1991;25:281–286.
4. Cockburn I. Assessment of the risks of malignancy and lymphoma developing in patients using Sandimmune. Transplant Proc. 1987;19:1804–1807.
5. Ahmed AR, Hombal SM. Cyclophosphamide (Cytoxan): a review on relevant pharmacology and clinical uses. J Am Acad Dermatol. 1984;11(6):1115–1126.
6. Roenigk HH Jr, Auerbach R, Maibach H, Weinstein G, Lebwohl M. Methotrexate in psoriasis: consenses conference. J Am Acad Dermatol. 1998;38:478–485.
7. Schreiber SL, Crabtree GR. The mechanism of action of cyclosporine and FK506. Immunol Today. 1992;13:136–142.
8. Lebwohl M, Ellis C, Gottlieb A, et al. Cyclosporine consensus conference: with an emphasis on the treatment of psoriasis. J Am Acad Dermatol. 1998;39:464–475.
9. Coleman MD. Dapsone toxicity: some current perspectives. Gen Pharmacol. 1995;26:1461–1467.
10. Coleman MD. Dapsone: modes of action, toxicity and possible strategies for increasing patient tolerance. Br J Dermatol. 1993;129:507–513.
11. Prussick R, Shear NH. Dapsone hypersensitivity syndrome. J Am Acad Dermatol. 1996;35:346–349.
12. Chaidemenos GC. Tetracycline and niacinamide in the treatment of blistering skin diseases. Clin Dermatol. 2001;19(6):781–785.
13. Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345(10):747–755.
14. Prins C, Gelfand EW, French LE. Intravenous immunoglobulin: properties, mode of action and practical use in dermatology. Acta Derm Venereol. 2007;87(3):206–218.
15. Czernik A, Bystryn JC. Improvement of intravenous immunoglobulin therapy for bullous pemphigoid by adding immunosuppressive agents: marked improvement in depletion of circulating autoantibodies. Arch Dermatol. 2008;144(5):658–661.
16. Cianchini G, Corona R, Frezzolini A, et al. Treatment of severe pemphigus with rituximab: report of 12 cases and a review of the literature. Arch Dermatol. 2007;143(8):1033–1038.
17. Rosenberg FR, Sanders S, Nelson CT. Pemphigus: a 20-year review of 107 patients treated with corticosteroids. Arch Dermatol. 1976;112(7): 962–970.
18. Werth VP. Treatment of pemphigus vulgaris with brief, high-dose intravenous glucocorticoids. Arch Dermatol. 1996;132(12):1435–1439.
19. Chams-Davatchi C, Esmaili N, Daneshpazhooh M, et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J Am Acad Dermatol. 2007;57(4):622–628.
20. Bredlich RO, Grundmann-Kollmann M, Behrens S, Kerscher M, Peter RU. Mycophenolate mofetil monotherapy for pemphigus vulgaris. Br J Dermatol. 1999;141(5):934.
21. Grundmann-Kollmann M., Kaskel P, Leiter U, et al. Treatment of pemphigus vulgaris and bullous pemphigoid with mycophenolate mofetil monotherapy. Arch Dermatol. 1999;135(6):724–725.
22. Lebwohl M, Coulson I, Heymann W, Berth-Jones J. Treatment of Skin Disease: Comprehensive Therapeutic Strategies. 2nd ed. 2005, New York: Mosby, Inc.
23. Czernik A, Beutner EH, Bystryn JC. Intravenous immunoglobulin selectively decreases circulating autoantibodies in pemphigus. J Am Acad Dermatol. 2008;58(5): 796–801.
24. El Tal AK, Posner MR, Spigelman Z, Ahmed AR. Rituximab: a monoclonal antibody to CD20 used in the treatment of pemphigus vulgaris. J Am Acad Dermatol. 2006. 55(3):449–459.
25. Pasricha JS, Khaitan BK, Raman RS, Chandra M. Dexamethasone-cyclophosphamide pulse therapy for pemphigus. Int J Dermatol. 1995;34(12):875–882.
26. Heaphy MR, Albrecht J, Werth VP. Dapsone as a glucocorticoid-sparing agent in maintenance-phase pemphigus vulgaris. 2005;141(6):699–702.
27. Pandya AG, Dyke C. Treatment of pemphigus with gold. Arch Dermatol. 1998;134(9):1104–1107.
28. Yeh SW, Ahmed B, Sami N, Razzaque Ahmed A. Blistering disorders: diagnosis and treatment. Dermatol Ther. 2003;16(3):214–223.
29. Mutasim DF, Adams BB, Immunofluorescence in dermatology. J Am Acad Dermatol. 2001. 45(6):803–822; quiz 822–824.
30. Martínez De Pablo MI, Iranzo P, Mascaró JM, et al. Paraneoplastic pemphigus associated with non-Hodgkin B-cell lymphoma and good response to prednisone. Acta Derm Venereol. 2005;85(3):233–235.
31. Borradori L, Lombardi T, Samson J, et al. Anti-CD20 monoclonal antibody (rituximab) for refractory erosive stomatitis secondary to CD20(+) follicular lymphoma-associated paraneoplastic pemphigus. Arch Dermatol. 2001; 137(3):269–272.
32. Vecchietti G, Kerl K, Hügli A, Samson J, Borradori L. Topical tacrolimus (FK506) for relapsing erosive stomatitis in paraneoplastic pemphigus. Br J Dermatol. 2003; 148(4):833–834.
33. Lin R, Ladd DJ Jr, Powell DJ, Way BV. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004; 140(7):889–890.
34. Mimouni D, Anhalt GJ, Cummins DL, et al. Treatment of pemphigus vulgaris and pemphigus foliaceus with mycophenolate mofetil. Arch Dermatol, 2003;139(6):739–742.
35. Gubinelli E, Bergamo F, Didona, et al. Pemphigus foliaceus treated with etanercept. J Am Acad Dermatol. 2006;55(6):1107–1108.
36. Fivenson DP, Breneman DL, Rosen GB, et al. Nicotinamide and tetracycline therapy of bullous pemphigoid. Arch Dermatol. 1994;130(6):753–758.
37. Beissert S, Werfel T, Frieling U, et al. A comparison of oral methylprednisolone plus azathioprine or mycophenolate mofetil for the treatment of bullous pemphigoid. Arch Dermatol. 2007;143(12):1536–1542.
38. Kjellman P, Eriksson H, Berg P. A retrospective analysis of patients with bullous pemphigoid treated with methotrexate. Arch Dermatol. 2008;144(5):612–616.
39. Gurcan HM, Ahmed AR, Frequency of adverse events associated with intravenous immunoglobulin therapy in patients with pemphigus or pemphigoid. Ann Pharmacother. 2007;41(10):1604–1610.
40. Smith EP, Zone JJ. Dermatitis herpetiformis and linear IgA bullous dermatosis. Dermatol Clin. 1993;11(3):511–526.
41. Garioch JJ, Lewis HM, Sargent SA, Leonard JN, Fry L. 25 years’ experience of a gluten-free diet in the treatment of dermatitis herpetiformis. Br J Dermatol. 1994;131(4):541–545.
42. Bernstein JE, Lorincz AL. Sulfonamides and sulfones in dermatologic therapy. Int J Dermatol. 1981;20(2):81–88.
43. Shah SA, Ormerod AD. Dermatitis herpetiformis effectively treated with heparin, tetracycline and nicotinamide. Clin Exp Dermatol. 2000;25(3): 204–205.
44. Stenveld HJ, Starink TM, van Joost T, Stoof TJ. Efficacy of cyclosporine in two patients with dermatitis herpetiformis resistant to conventional therapy. J Am Acad Dermatol. 1993;28(6):1014–1015.
45. Lang PG Jr. Dermatitis herpetiformis responsive to systemic corticosteroids. J Am Acad Dermatol. 1985;13(3): 513–515.
46. Silvers DN, Juhlin EA, Berczeller PH, McSorley J. Treatment of dermatitis herpetiformis with colchicine. Arch Dermatol. 1980; 116(12):1373–1384.
47. Ang P, Tay YK. Treatment of linear IgA bullous dermatosis of childhood with colchicine. Pediatr Dermatol. 1999;16(1):50–52.
48. Yomada M, Komai A, Hashimato T. Sublamina densa-type linear IgA bullous dermatosis successfully treated with oral tetracycline and niacianamide. Br J Dermatol. 1999;141(3):608–609.
49. Alajlan A, Al-Khawajah M, Al-Sheikh O, et al. Treatment of linear IgA bullous dermatosis of childhood with flucloxacillin. J Am Acad Dermatol. 2006;54(4):652–656.
50. Segura S, Iranzo P, Martínez-de Pablo I, et al. High-dose intravenous immuno-globulins for the treatment of autoimmune mucocutaneous blistering diseases: evaluation of its use in 19 cases. J Am Acad Dermatol. 2007; 56(6):960–967.
51. Al-Fouzan AW, Galadari I, Oumeish I, Oumeish OY. Herpes gestationis (Pemphigoid gestationis). Clin Dermatol. 2006;24(2):109–112.
52. Van de Wiel A, Hart HC, Flinterman J, et al. Plasma exchange in herpes gestationis. Br Med J. 1980;281(6): 1041–1042.
53. Kreuter A, Harati A, Breuckmann F, Appelhans C, Altmeyer P. Intravenous immune globulin in the treatment of persistent pemphigoid gestationis. J Am Acad Dermatol. 2004;51(6): 1027–1028.
54. Rodrigues Cdos S, Filipe P, Solana Mdel M, et al. Persistent herpes gestationis treated with high-dose intravenous immunoglobulin. Acta Derm Venereol. 2007;87(2):184–186.
55. Loo WJ, Dean D, Wojnarowska F. A severe persistent case of recurrent pemphigoid gestationis successfully treated with minocycline and nicotinamide. Clin Exp Dermatol. 2001;26(8):726–727.
56. Cunningham BB, Kirchmann TT, Woodley D. Colchicine for epidermolysis bullosa acquisita. J Am Acad Dermatol. 1996;34(5 Pt 1):781–784.
57. Schmidt E, Benoit S, Bröcker EB, Zillikens D, Goebeler M. Successful adjuvant treatment of recalcitrant epidermolysis bullosa acquisita with anti-CD20 antibody rituximab. Arch Dermatol. 2006; 142(2):147–150.