
James Q. Del Rosso, DO, FAOCD, Dermatology Residency Director, Valley Hospital Medical Center, Las Vegas, Nevada
Sanjay Bhambri, DO, Chief Dermatology Resident (PGY-4), Valley Hospital Medical Center, Las Vegas, Nevada
Abstract
Objective: To assess the efficacy and safety of topical fluocinonide 0.1% cream for the treatment of atopic dermatitis. Design: In this double-blind, vehicle-controlled study, patients were randomized to receive treatment with fluocinonide 0.1% cream applied once (n=109) or twice daily (n=102) or vehicle applied once (n=50) or twice daily (n=52) for two weeks. Setting: Multicenter, outpatient. Participants: Patients aged 18 years or older with atopic dermatitis affecting at least two percent but less than 10 percent of body surface area. Measurements: Efficacy and safety measures included lesion severity, pruritus, hypothalamic-pituitary-adrenal axis suppression, and adverse events. Results: Fluocinonide 0.1% cream applied once or twice daily was more effective than cream vehicle. Both regimens were similarly efficacious after two weeks of treatment. At the end of treatment, lesions were cleared or almost cleared in 59 percent of subjects treated once daily and 57 percent of subjects treated twice daily with fluocinonide 0.1% cream. Further, considerable residual benefit remained after cessation of twice-daily versus once-daily treatment. Skin safety evaluations showed no significant adverse effects of treatment on signs or symptoms of skin atrophy. Fluocinonide 0.1% cream and vehicle treatments did not differ significantly in their suppression of the hypothalamic-pituitary-adrenal axis, nor did hypothalamic-pituitary-adrenal axis suppression differ significantly following once- or twice-daily treatment with fluocinonide 0.1% cream. Fluocinonide 0.1% cream was well tolerated. Conclusion: Once- or twice-daily topical application of fluocinonide 0.1% cream for 14 days was safe and effective for treating atopic dermatitis in this adult patient population. The efficacy of once-daily application was comparable to twice-daily application.
(J Clin Aesthetic Dermatol. 2009;2(9):24–32.)
Atopic dermatitis is a chronic disease with acute flare-ups and periods of remission. It is often the first manifestation of a group of allergic disorders that includes asthma, allergic rhinitis, and food allergy.[1,2] Both genetic and environmental factors influence development of the disease.[3] Approximately 10 to 20 percent of children and 1 to 3 percent of adults are diagnosed with atopic dermatitis.[2] The disease develops during the first 12 months of life in 75 percent of children who are affected and clears completely at, or shortly after, puberty in 40 to 60 percent[4,5]; however, atopic dermatitis may persist in more than 20 percent of adolescents, and up to 17 percent of adults may experience the onset of atopic dermatitis after adolescence.[6,7]
The clinical presentation of atopic dermatitis varies with age and disease activity.[2,5,8,9] In children between two years of age and puberty, the exudative lesions observed in infancy are less common.[2,9] Rather, lesions tend to be more chronic and characterized by severe pruritus with excoriations and lichenified papules and plaques and involve the hands, feet, wrists, ankles, and antecubital and popliteal regions.[2,5] After puberty and continuing into adulthood, main areas of involvement include the flexural folds; face and neck; upper arms and back; and the dorsa of the hands, feet, fingers, and toes.[2,5] Dry, scaling, erythematous papules and plaques are characteristic, and large lichenified plaques are formed as the lesions become more chronic.[2,5] Although weeping, crusting, and exudation may occur, these findings are usually due to staphylococcal skin infection.[5]
Managing atopic dermatitis requires a multifactorial approach that includes regular use of emollients and skin hydration, antipruritic therapy, topical anti-inflammatory agents, identification and elimination of possible triggers, and systemic antibiotic treatment if widespread secondary bacterial infection, mainly due to Staphylococcus aureus, is present.[2,9] For more than 40 years, topical corticosteroids have been a key component of the management of atopic dermatitis[10] and continue to be important agents, particularly for the control of acute flare-ups.[9] In addition to their anti-inflammatory effect, the use of topical steroids reduces skin colonization with S. aureus.[9,11]
Topical corticosteroid formulations ranging from low potency to super potency are available.[5] Recent guidelines recommend using low- to mid-potency topical corticosteroids for the treatment of mild-to-moderate atopic dermatitis and more potent preparations for moderate-to-severe disease and for patients with lichenified plaques.[9] The general rule has been to use the least potent, most effective topical corticosteroid for the treatment of atopic dermatitis. A possible disadvantage of using a lower-potency preparation initially is that this may fail to improve or even worsen the disease, resulting in poor treatment adherence.[12] An emerging strategy is to “hit hard and hit fast” by initiating treatment with a mid- or high-potency topical corticosteroid (except for lesions on the face, axillae, or groin) to gain rapid control of the disease, and then maintain improvement with a low-potency preparation, or to use a potent preparation intermittently (e.g., twice weekly or two consecutive days each week) to reduce flares.[12,13]
The optimal dosage frequency of topical corticosteroids in atopic dermatitis has not been fully evaluated and remains unclear[14,15]; however, twice-daily (BID) application, the most commonly recommended regimen based on approved product labeling of many topical corticosteroids, has evolved empirically over time.[14,15] Frequency of application is an important clinical consideration, as it is likely to be a major factor influencing patient adherence to treatment. Data from 76 studies indicate that the rate of adherence with BID dosing is lower than with once-daily (QD) dosing (69% vs. 79%).[16] A recent systematic review of the literature comparing QD versus BID application of topical corticosteroids identified only seven randomized, controlled trials of the same active compound and three comparing different agents of the same potency that met the inclusion criteria of the review.[14] It was concluded that the efficacy of the same potency topical corticosteroids used QD and BID was similar and did not favor one regimen over another[14]; however, an earlier, similar systematic review concluded that, in the absence of clear evidence from randomized, controlled trials to support BID over QD topical corticosteroid application, initiating therapy in all patients with a QD regimen would be reasonable.[10] It was also noted that the vehicle used in the topical formulation may enhance efficacy, and that when two treatments are equally effective, the cosmetic acceptability of one vehicle over another may facilitate long-term use.[10]
Fluocinonide 0.1% cream, a super-potent topical corticosteroid, is currently available in the United States. Although this formulation contains a two-fold higher concentration of fluocinonide than what is found in earlier products (0.05%), the vehicle characteristic of the cream allows for marked retention of the active ingredient in the stratum corneum, epidermis, and dermis, with a lesser propensity for systemic absorption.[17] Additionally, the cream vehicle is similar to a conventional ointment base in that its water content is very low (<1%); however, it is devoid of the greasiness of an ointment.[17] Similar to the other available super-potent topical corticosteroids, with the exception of clobetasol propionate 0.05% formulations, fluocinonide 0.1% is approved for QD or BID application.[18]
The following Phase 3 clinical trial was undertaken to evaluate the efficacy and safety of fluocinonide 0.1% cream compared to vehicle when applied topically QD or BID for two weeks for the treatment of adults with atopic dermatitis.
MATERIALS AND METHODS
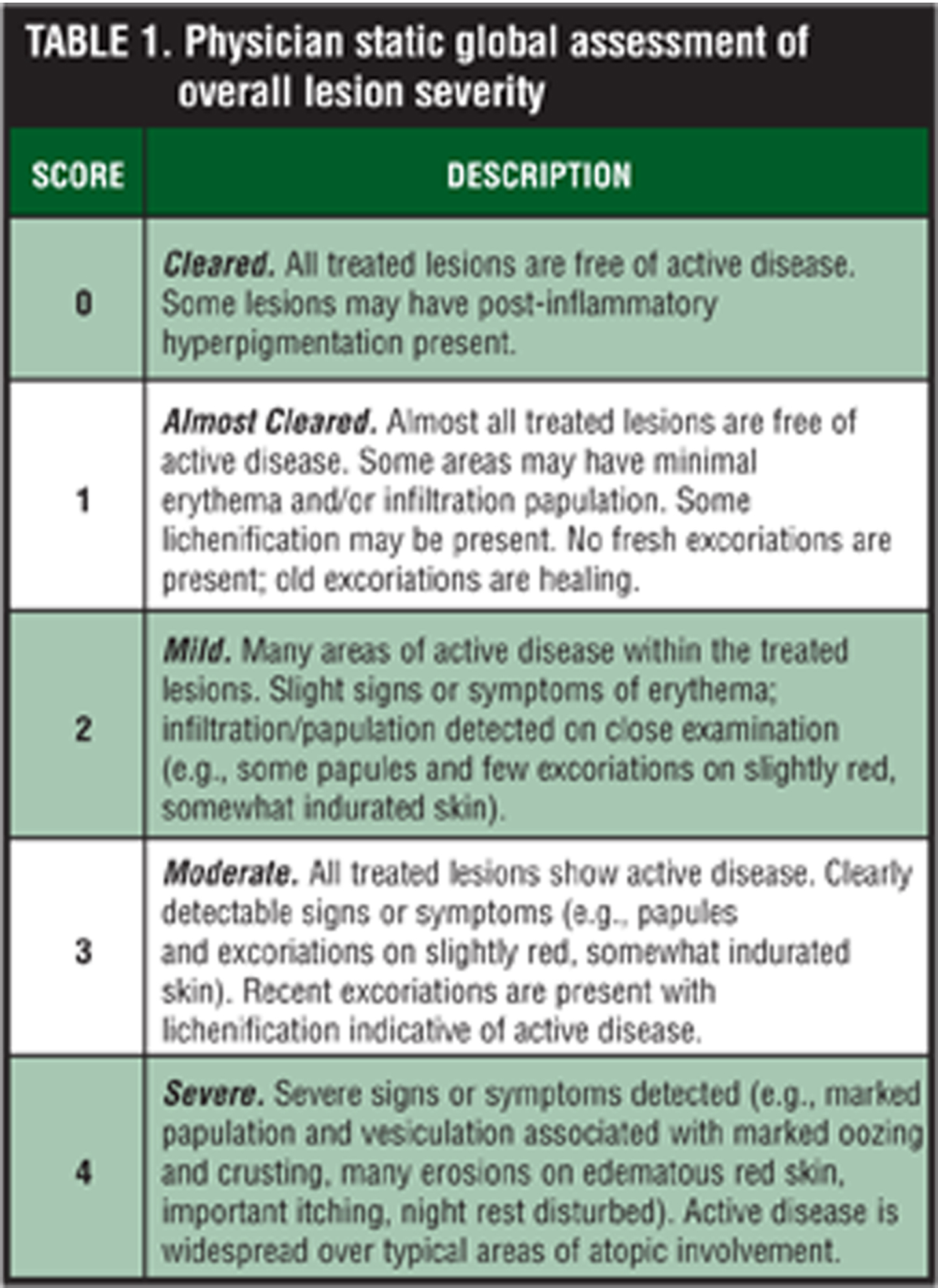
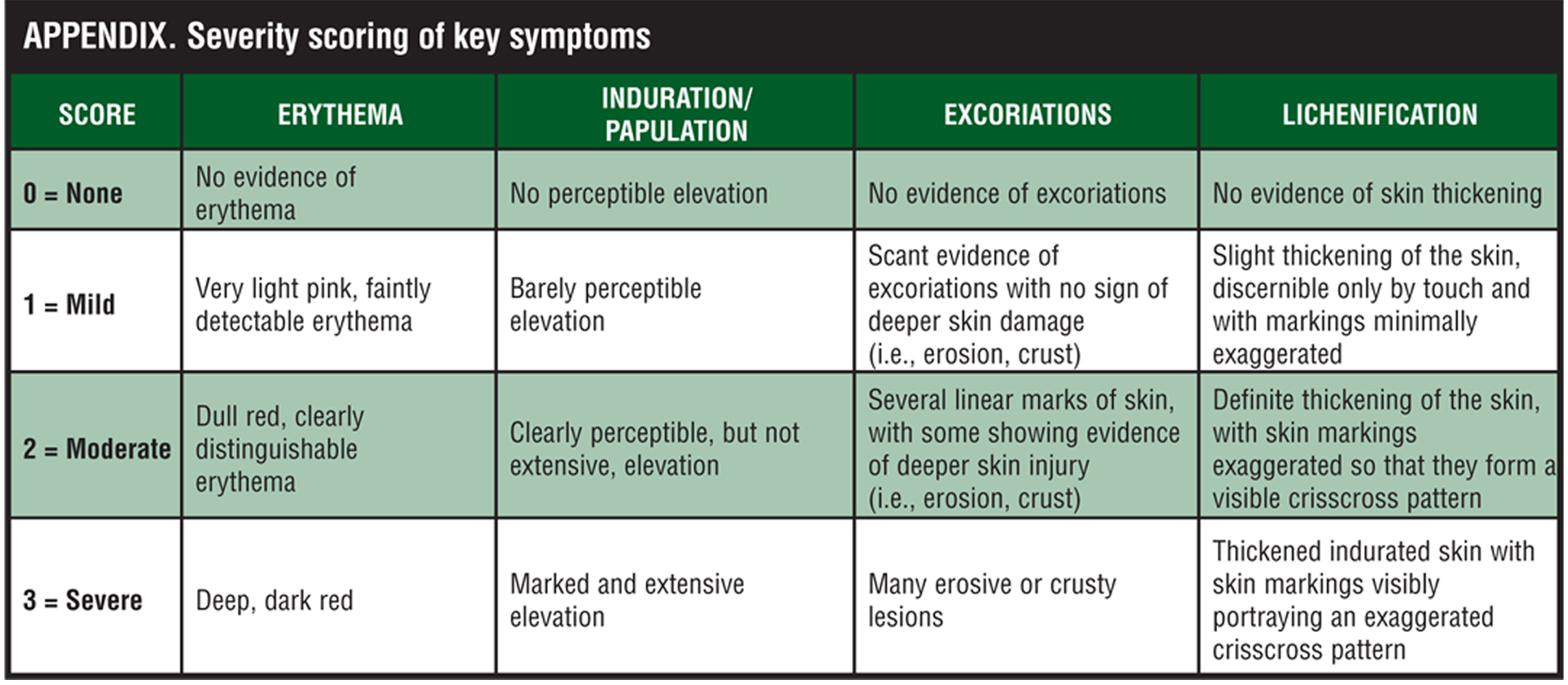
Subjects. Otherwise healthy subjects 18 years of age or older with clinically diagnosed atopic dermatitis with a treatable area involving at least two percent but no more than 10 percent of body surface area (BSA), determined by the investigator using the “rule of nines,” and who had been clinically stable for at least one month were enrolled in the study.[19] Eligible subjects had a minimum total symptom score of 7 out of 12 based on the investigator’s evaluation (on a scale from 0=none to 3=severe) of the severity of (a) erythema, (b) infiltration/papulation, (c) excoriations, and (d) lichenification of all affected treatable areas, at least mild pruritus (score ?1), and a physician global assessment of overall lesion severity of at least 3 (moderate). Subjects were free of any systemic or dermatological disorder that might interfere with the study results or increase the risk of adverse events.
Excluded from the study were subjects who had not undergone a washout period of two weeks for topical corticosteroids, topical retinoids, or topical calcineurin inhibitors; four weeks for systemic corticosteroids, systemic retinoids, or prolonged sun exposure or ultraviolet light therapy; and 16 weeks for systemic immunomodulating biological agents, such as etanercept. Also excluded were subjects with unstable atopic dermatitis (spontaneously improving or worsening) and those with any untreated bacterial, mycobacterial, fungal, or viral skin lesion. A washout period of four weeks for any medication known to affect serum cortisol levels or hypothalamic-pituitary-adrenal (HPA) axis function was required for subjects undergoing HPA evaluation. Patients with irregular sleep schedules or those who worked night shifts were excluded from HPA evaluation due to the physiological diurnal variation of cortisol levels.
Study design. This Phase 3, double-blind, randomized, parallel-group, vehicle-controlled study was conducted at 25 centers in the United States and was approved by the Institutional Review Board at each participating center. During the baseline visit, subjects were randomized to receive either fluocinonide 0.1% cream or its vehicle. Half of the subjects were randomly selected to apply the cream either QD in the morning or evening, and half were instructed to apply the cream BID, morning and evening, for 14 consecutive days to all affected, treatable areas of the skin.
For subjects enrolled at sites performing the cosyntropin stimulation test (CST), pre-stimulation cortisol levels followed by cosyntropin injection and then post-stimulation cortisol blood levels were obtained at baseline. After the baseline visit, patients returned after treatment Weeks 1 and 2 and at two weeks post-treatment (Week 4) for evaluations of efficacy, skin safety, and to obtain information on adverse events and use of concomitant medications. At Week 4, subjects who had a normal CST at baseline, but abnormal results at the end of treatment (Week 2), underwent an additional CST.
Efficacy assessments. At each study visit, the investigator rated overall lesion severity on a 5-point scale considering all of the treatable lesions designated at the baseline visit (Table 1). The severity of erythema, infiltration/papulation, excoriations, and lichenification of all treatable lesions was scored independent of previous assessments on a 4-point scale where 0=none to 3=severe. For the severity scoring of these key symptoms, refer to the Appendix. Overall severity of pruritus was rated as 0=none (no itching), 1=mild (slightly bothersome itching), 2=moderate (bothersome itching, but no loss of sleep), and 3=severe (constant itching causing intense discomfort and loss of sleep). The extent of involvement of atopic dermatitis was assessed at each visit in terms of the total BSA, calculated using the “rule of nines” and recorded on a full-body diagram.[19]
{kind=link}
{kind=link}
Safety assessments. Skin safety evaluations of all treated lesions were made at each study visit by rating the following seven signs and symptoms of skin atrophy as present or absent: telangiectasis, skin transparency, loss of elasticity, loss of normal skin markings, skin thinning, striae, and bruising. All local and systemic adverse events were recorded at each study visit.
HPA axis suppression was assessed at selected sites using the CST. Testing occurred at the baseline visit, prior to application of study medication, and at the end of treatment (Week 2). Subjects with a normal CST at baseline, but abnormal results at Week 2, were re-tested at Week 4 (two weeks post-treatment). Those with abnormal results at baseline and the end of treatment could be re-tested at the investigator’s discretion. On each test day, pre-stimulation blood samples were obtained between 6:30 and 8:30 a.m. prior to intravenous injection of 0.25mg of cosyntropin and application of study medication. Samples were collected again 30 minutes after injection. HPA axis suppression was defined as a basal serum cortisol level (pre-stimulation) less than or equal to 5µg/dL, or a 30-minute post-stimulation level less than or equal to18µg/dL, or a post-stimulation increase over the basal level <7 µg/dL.
Statistical analysis. Demographic data, background characteristics of subjects, and adverse events were summarized for each treatment group using the intent-to-treat population consisting of 313 enrolled subjects. Frequency counts were presented for the incidence of signs and symptoms of skin atrophy by treatment group. CST results were summarized by treatment group. The primary efficacy parameter was the dichotomized physician static global assessment (PGA) of overall lesion severity at Week 2 (end of treatment), with treatment success defined as a score of 0 (cleared) or 1 (almost cleared). This parameter was compared between each active treatment regimen and its vehicle control (2 comparisons) and between the two active and the two vehicle treatments (2 comparisons) using pairwise Mantel-Haenszel statistics, stratified on investigational site. For this study, there is 80-percent power to detect a 20-percent difference between the two active treatment arms. To control for Type I error, the QD regimen was tested against its vehicle only if the BID regimen test against its vehicle was statistically significant. The comparison between the two active treatments was tested only if both comparisons with vehicle were statistically significant. The Cochran-Mantel-Haenszel row mean score statistic was used to analyze PGA, individual symptom scores, total symptom score, overall severity of pruritus, and BSA of affected areas at each visit, stratifying on investigational site. The extent of rebound, a secondary endpoint, was evaluated based on the comparison of symptom scores and PGA between treatments at Week 4 (two weeks post-treatment).
RESULTS
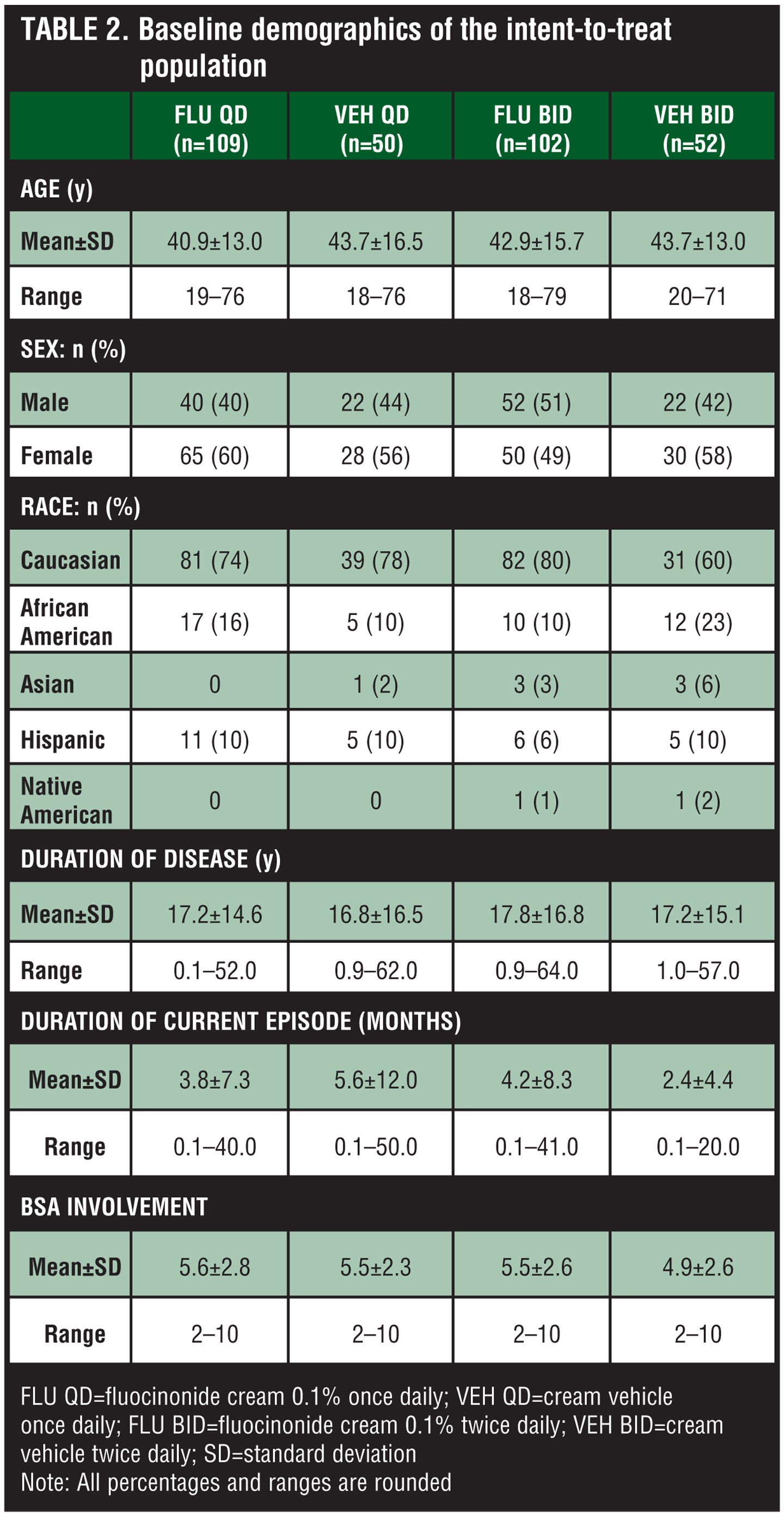
Study population. Three hundred thirteen subjects were enrolled in the study and randomized to treatment with fluocinonide 0.1% QD (n=109), fluocinonide 0.1% BID (n=102), control vehicle QD (n=50), and control vehicle BID (n=52). Demographic and baseline characteristics were similar between treatment groups (Table 2). Subjects ranged in age from 18 to 79 years (mean age approximately 41–44 years), most (greater than or equal to 60%) of whom were Caucasian, and with the exception of the fluocinonide 0.1% BID group, most were female. The mean duration of atopic dermatitis was 17 to 18 years, and the average duration of the current episode was 2.4 to 5.6 months. The mean percentage of BSA involvement ranged from 4.9 to 5.6 percent.
{kind=link}
Concomitant medications were taken by 229 subjects: 81 (74%) in the fluocinonide 0.1% QD group, 74 (73%) in the fluocinonide 0.1% BID group, 35 (70%) in the vehicle QD control group, and 39 (75%) in the vehicle BID control group. The most common classes of agents were anti-inflammatory and anti-rheumatic agents, oral antihistamines, hormones for oral contraception or replacement therapy, analgesics, vitamins, anti-asthmatic agents, oral antibiotics, and psychoanaleptic agents. Topical corticosteroids other than study medication were used by four patients in the fluocinonide 0.1% QD group (fluocinonide, hydrocortisone, nystatin-triamcinolone, and triamcinolone; 1 patient each) and one patient in the vehicle QD control group (diflorasone diacetate). During the treatment phase, one patient in the fluocinonide 0.1% QD group continued use of nystatin-triamcinolone. This subject was included in the intent-to-treat (ITT) analysis.
Twenty-two subjects discontinued the study for the following reasons: adverse events (n=5; 1.6%), protocol violation (n=1; 0.3%), subject’s request (n=5; 1.6%), lost to follow up (n=9; 2.9%), and other reasons (n=2; 0.6%). Adverse events leading to discontinuation from the study were worsening of atopic dermatitis (1 subject in each active treatment group and 1 subject in the vehicle BID control group), skin fissures, bleeding, peeling, and severe atopic dermatitis in two subjects in the vehicle QD control group.
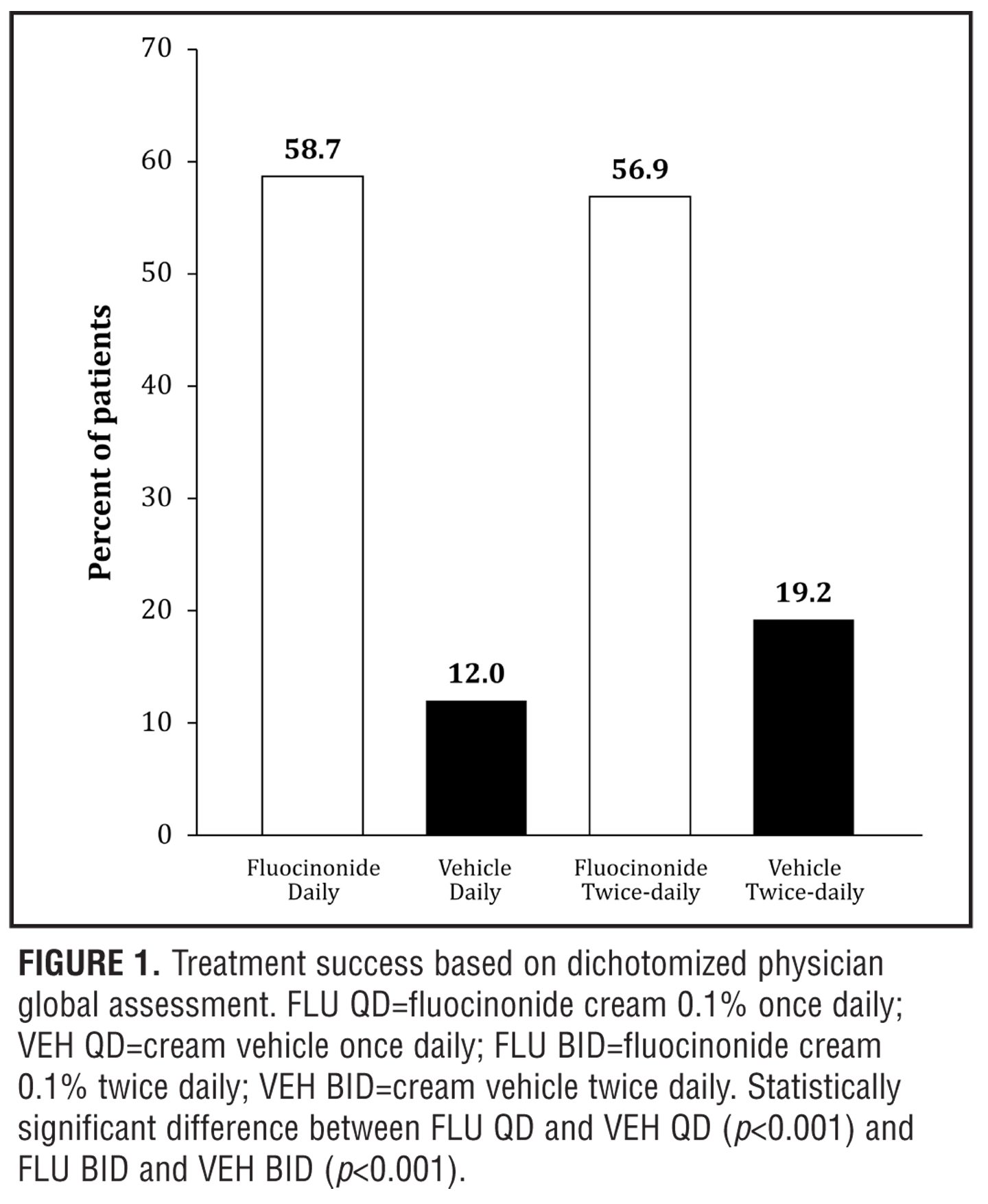
Efficacy results. Both fluocinonide 0.1% QD and fluocinonide 0.1% BID treatments were superior to vehicle and similarly efficacious in the rate of treatment success (defined as lesions cleared or almost cleared; Figure 1) after two weeks of active treatment. At the end of the two-week treatment period, fluocinonide 0.1% cream applied either QD or BID significantly improved PGA scores compared to the corresponding vehicle control (Figure 1; p<0.001). Treatment success, defined as a score of 0 (cleared) or 1 (almost cleared), was achieved for 59 percent (64/109) of subjects in the fluocinonide 0.1% QD group and 57 percent (58/102) of subjects in the fluocinonide 0.1% BID group compared to 12 percent (6/50) of subjects in the vehicle QD control group and 19 percent (10/52) of subjects in the vehicle BID control group. Efficacy was significantly greater for both the QD and BID 0.1% fluocinonide groups compared to vehicle (p<0.001). By Week 4 (i.e., two weeks after the last dose of study medication), criteria for treatment success continued to be met by 30 percent (33/109) and 47 percent (48/102) of subjects in the fluocinonide 0.1% QD and fluocinonide 0.1% BID groups, respectively, compared to 18 percent (9/50) and 27 percent (14/52) of subjects in the vehicle QD and vehicle BID control groups, respectively.
{kind=link}
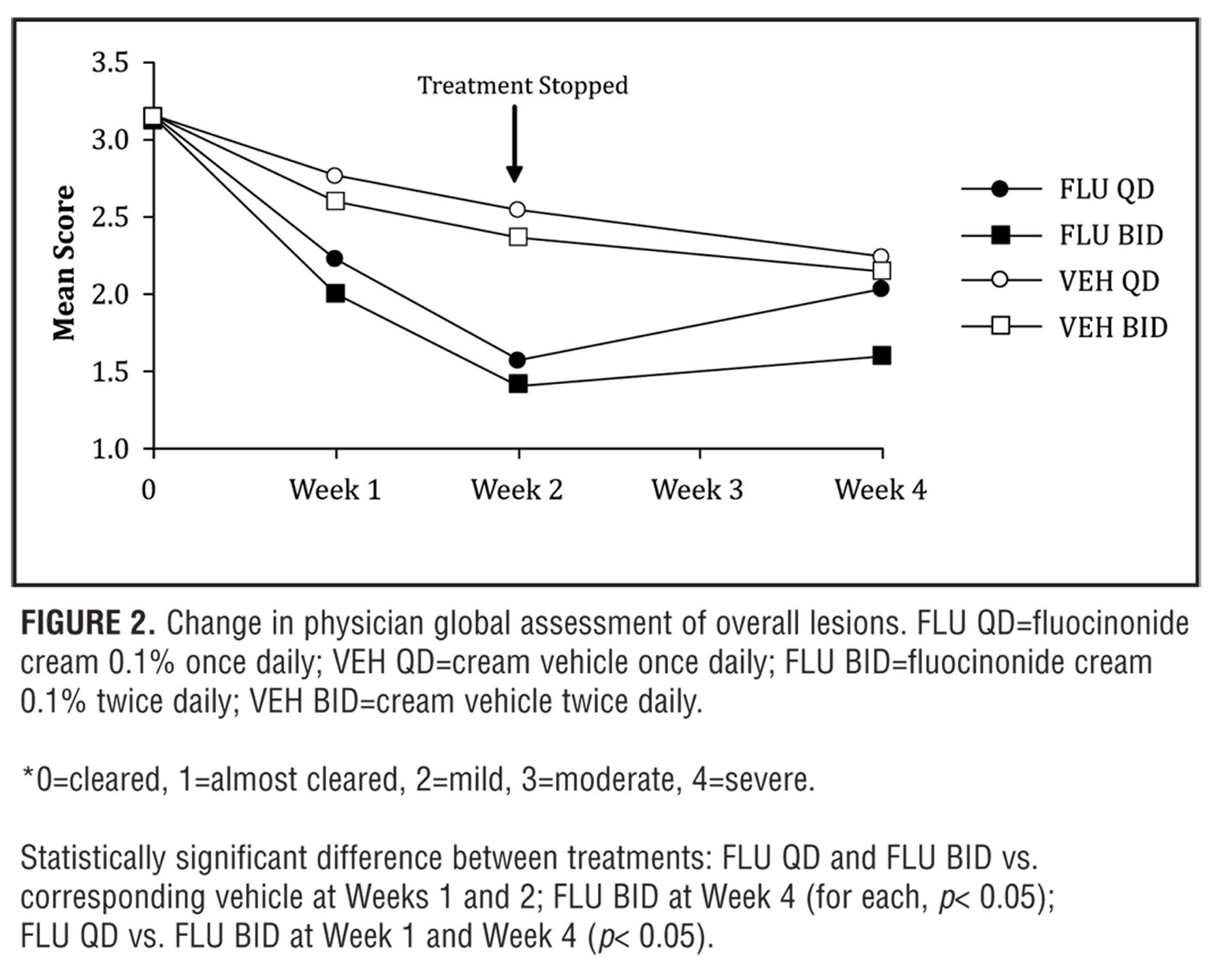
Efficacy data in Figure 2 shows the Physician Ratings of Overall Lesion Severity. At the end of treatment, the responses in the fluocinonide 0.1% treatment groups were significantly greater than those in the corresponding vehicle control groups (p<0.05). The apparent increased response with fluocinonide 0.1% BID versus fluocinonide 0.1% QD was not statistically significant. A greater residual treatment effect was seen in the fluocinonide 0.1% BID group two weeks after the end of treatment compared to fluocinonide 0.1% QD (Week 4) (p<0.05).
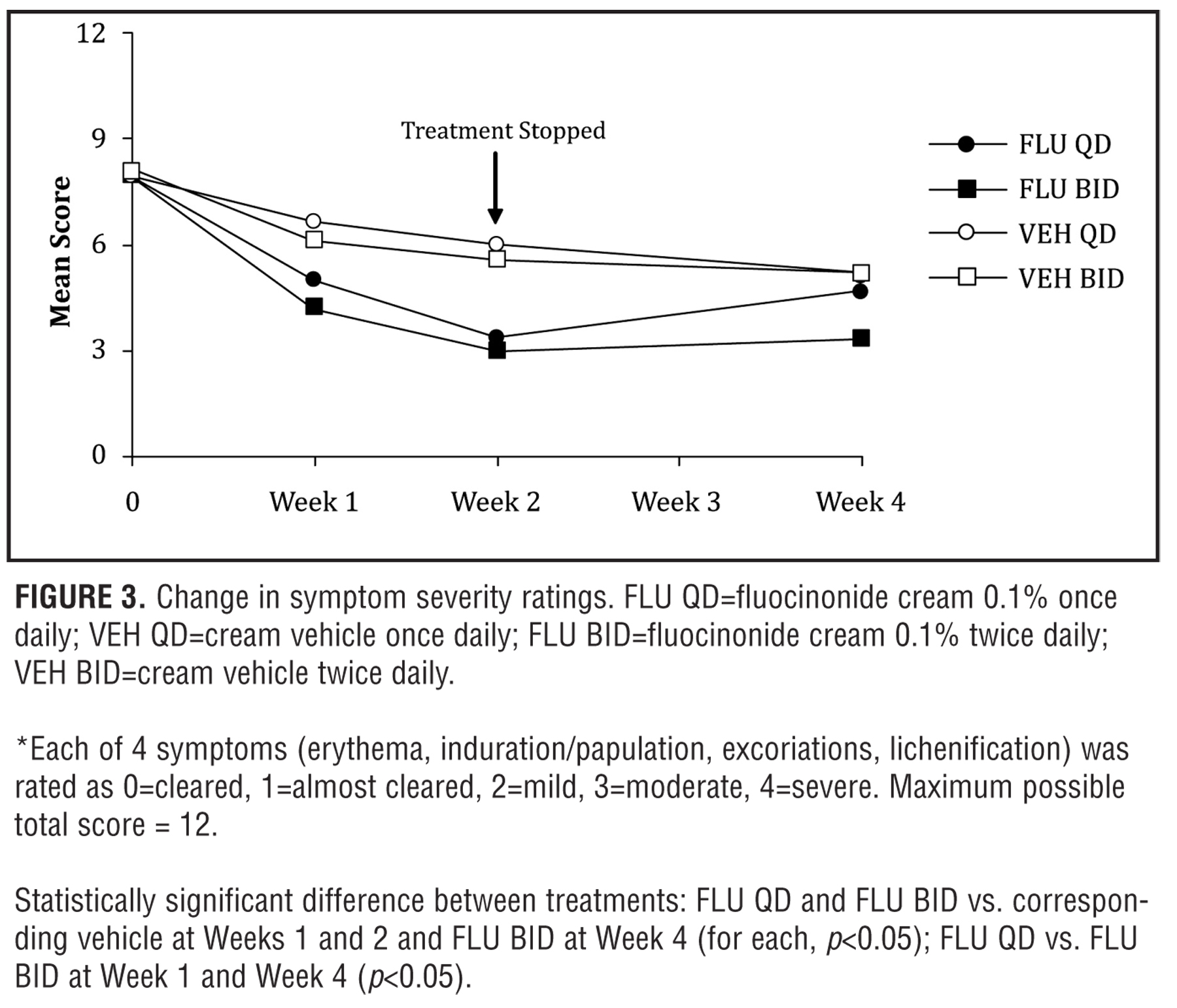
The overall ratings for the symptoms of erythema, induration/papulation, excoriations, and lichenification produced total symptom scores that were significantly decreased at Week 2 in both active treatment groups compared to corresponding vehicle control groups (p<0.05) (Figure 3). A residual reduction in all symptoms with the exception of erythema was seen at Week 4 in the fluocinonide 0.1% BID versus fluocinonide 0.1% QD group (p<0.05). Improvements also were observed in the subject overall ratings of pruritus, and the physician ratings of percent BSA affected at Week 2 for both fluocinonide 0.1% QD and fluocinonide 0.1% BID groups compared with their respective vehicle control groups (both treatments, p<0.001 for pruritus and p?0.002 for %BSA).
{kind=link}
{kind=link}
Thus, overall, disease severity was reduced significantly for both QD and BID active treatment groups, with maximal improvements seen at Week 2. Interestingly, after treatment stopped at the end of Week 2, the efficacy scores in the fluocinonide 0.1% QD group tended to regress toward those seen at Baseline from Week 2 to Week 4, while the efficacy scores in the fluocinonide 0.1% BID group regressed only slightly compared to the corresponding vehicle control group (p<0.05).
Safety assessments. Adverse events. Adverse events considered related to treatment were reported by 22 subjects: four (4%) in the fluocinonide 0.1% QD group, four (4%) in the fluocinonide 0.1% BID group, 10 (20%) in the vehicle QD control group, and four (8%) in the vehicle BID control group. The most common treatment-related adverse event was application-site burning, which was reported by two or more subjects in each treatment group. Application-site burning was reported 17 times by 15 subjects who described it as mild (n=12), moderate (n=3), and severe (n=2).
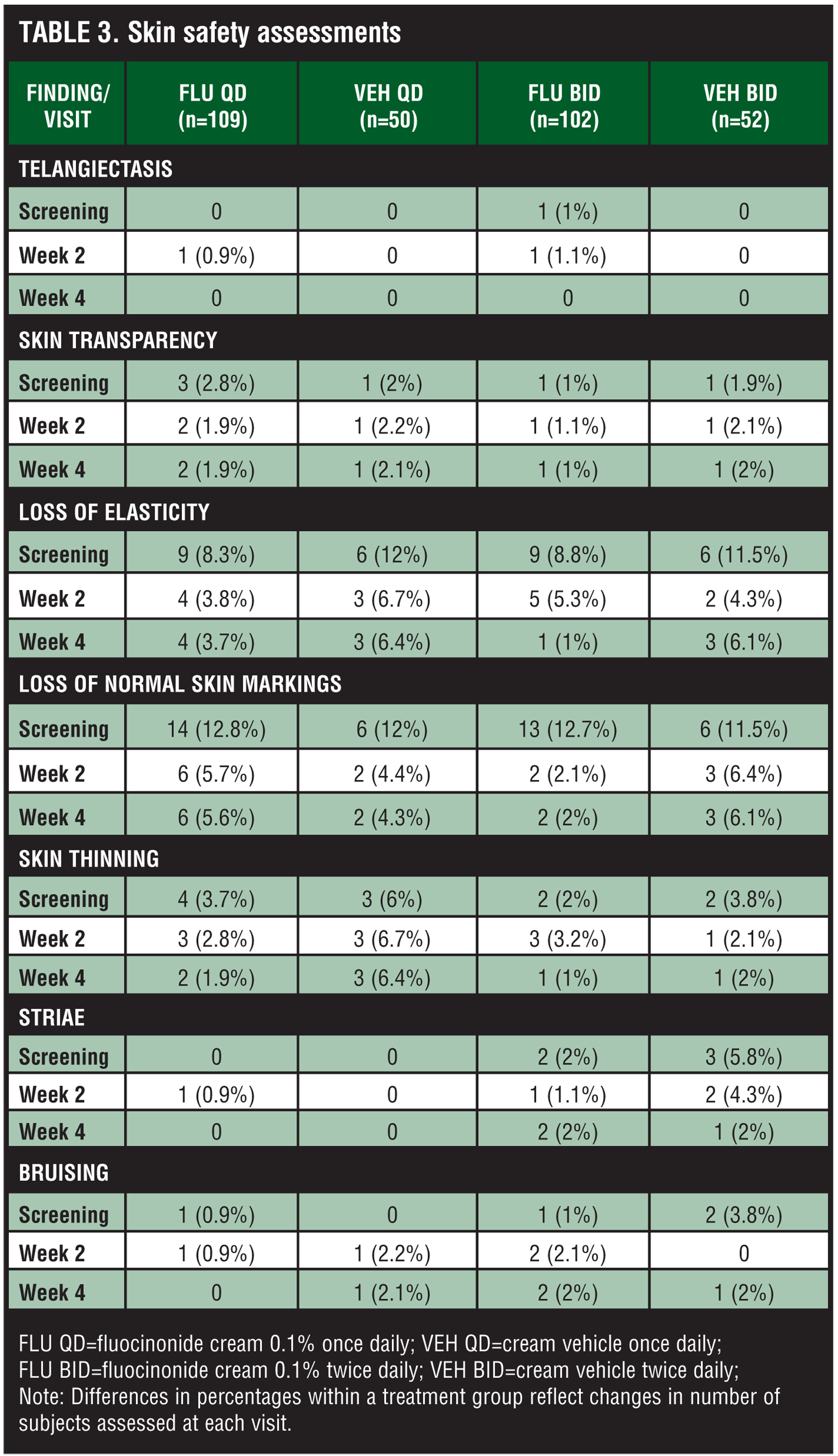
Skin safety evaluations. Skin safety evaluations showed no significant effects of treatment in signs and symptoms of skin atrophy (Table 3). Telangiectasis, skin transparency, loss of elasticity, loss of normal skin markings, skin thinning, striae, and bruising were absent in the majority of subjects, and no significant changes from baseline evaluations to the end of the study were observed.
{kind=link}
Adrenal responsiveness. CST was performed on 18 subjects in the fluocinonide 0.1% QD group, 15 subjects in the fluocinonide 0.1% BID group, and nine subjects each in the vehicle QD and vehicle BID control groups. No pretreatment CST data were available for one subject in the fluocinonide 0.1% BID group. At the screening/ baseline visit, 12 (24%) of 51 subjects (5 subjects in each fluocinonide 0.1% treatment group; 2 subjects in the vehicle QD group) met the criteria of HPA axis suppression. In these 12 subjects, no evidence of HPA axis suppression was found in 3 (60%) of 5 subjects in the fluocinonide 0.1% QD control group at Week 2 or a follow-up study visit. Of subjects with no HPA axis suppression at screening/baseline, HPA axis suppression following treatment with fluocinonide 0.1% cream was not significantly different from that following vehicle. HPA axis suppression following treatment with fluocinonide 0.1% BID was also not significantly different from that following QD treatment. HPA axis function returned to normal by the Week 4 follow-up visit in all subjects for whom repeat CST results were available: four subjects in the fluocinonide 0.1% QD group and one subject each in the fluocinonide 0.1% BID group and the vehicle QD control group.
The majority of subjects who met the criteria for HPA axis suppression did so due to a post-stimulation serum cortisol <7µg/dL over basal levels. In the fluocinonide 0.1% QD group, one subject had a pre-stimulation serum cortisol level less than or equal to 5µg/dL at the Week 2 visit, and one subject had a poststimulation serum cortisol level less than or equal to 18µg/dL at screening/baseline and at Week 2. Serum cortisol levels after CST at Week 2 were similar among fluocinonide 0.1% QD and fluocinonide 0.1% BID groups and vehicle QD and vehicle BID control groups, values being 10.8±8.5µg/dL, 10.8±4.6µg/dL, 11.0±4.7µg/dL, and 11.4±6.5µg/dL, respectively.
DISCUSSION
Results of this two-week, randomized, double-blind, multicenter study in adults with atopic dermatitis demonstrate that a once-daily regimen of fluocinonide 0.1% cream is as effective as a twice-daily regimen, and that both are more effective than the cream vehicle. Treatment success (lesions cleared or almost cleared) was achieved in most subjects treated with fluocinonide 0.1% cream QD or BID. Both regimens significantly decreased overall lesion severity, symptom severity ratings, total symptom ratings, and pruritus ratings at the end of treatment. Fluocinonide 0.1% cream was well tolerated; only four patients each in the QD and BID groups (only 8 out of 211 patients total) reported treatment-related adverse events, mainly mild or moderate application-site burning, all of which resolved.
Skin safety evaluations showed that QD and BID treatment with fluocinonide 0.1% cream was not associated with significant effects on skin integrity. Signs of skin atrophy, such as telangiectasis, striae, and bruising, were absent in the majority of subjects at the end of treatment. These findings are consistent with those of two studies that specifically assessed the skin atrophy potential of fluocinonide 0.1% cream.[20,21] In the first study, 20 subjects with steroid-responsive dermatoses were randomly assigned to apply fluocinonide 0.1% cream to disease-free target areas on one arm and clobetasol 0.5% cream to the other arm BID for two weeks.[20] Punch biopsies showed no significant reduction in epidermal thickness in fluocinonide-treated sites compared to a significant reduction in clobetasol-treated sites (p<0.0001). Sixteen of the 20 subjects did not experience any signs of cutaneous atrophy.[20] In a similar study, 10 female subjects applied four test preparations to specific sites on the forearms BID for 21 days: fluocinonide 0.1% cream, clobetasol propionate 0.05% cream, clobetasol propionate 0.05% foam, and the fluocinonide cream vehicle.[21] Evaluations of treated sites for skin changes (videomicroscopic and clinical), transepidermal water loss, epidermal thickness (by optical coherence tomography), and histological assessment of punch biopsies from treated sites consistently ranked the four test products as follows, in order of increasing atrophogenic potential: vehicle < fluocinonide 0.1% cream < clobetasol propionate 0.05% foam < clobetasol propionate 0.05% cream.[21]
Among subjects with a normal serum cortisol response to CST at screening/baseline, treatment with fluocinonide 0.1% cream and vehicle treatment did not differ significantly in HPA axis suppression nor did BID fluocinonide treatment differ significantly from QD treatment in HPA axis suppression. Thus, adopting QD dosing to specifically reduce the incidence of HPA suppression in this population of adults with atopic dermatitis does not appear necessary; however, as shown in this study of adult subjects, BID application of fluocinonide 0.1% cream does not improve efficacy in atopic dermatitis as compared to QD use. In a separate study of two-week fluocinonide 0.1% cream treatment in pediatric subjects with atopic dermatitis, there was no evidence of HPA axis suppression on CST testing in any subject with QD application, even in children <2 years of age.[22]
In addition to lower drug exposure, once-daily application may improve adherence and enhance therapeutic efficacy in patients of all ages. A meta-analysis of 11 studies of patients with skin disorders found that mean adherence to the prescribed treatment was 77 percent.[23] That adherence increases as the frequency of dosing decreases is well known.[16,24] A review of studies in which adherence was assessed using electronic monitoring devices found that adherence decreased from 79 percent with once-daily dosing to 69 percent with twice-daily dosing.[16] In the present clinical trial, the marginal benefit in efficacy (i.e., more rapid improvement, sustained post-treatment response) seen with twice-daily application of fluocinonide 0.1% cream compared to once-daily application needs to be weighed against the likelihood of decreased adherence to twice-daily dosing in actual use. In fact, the marginal increase in efficacy associated with twice-daily application of fluocinonide 0.1% cream that was documented using the standardized criteria mandated by the study protocol may not be observed or appreciated in “real world” clinical practice as the improvement in signs and symptoms of atopic dermatitis closely mirrored what was achieved with once-daily application.
CONCLUSION
Topical application of fluocinonide 0.1% cream applied once or twice daily for 14 days is safe and effective for treating atopic dermatitis in subjects aged 18 years and older. Once-daily applications provided the lowest effective dose of medication, while twice-daily application offered no differences in safety and marginal benefit in efficacy over once-daily applications.
Acknowledgment
The authors thank the members of the VANOS Study Group: William Abramovitz, MD, Dallas, TX; Melanie Appell, MD, Birmingham, AL; Suzanne Bruce, MD, Houston, TX; Steven A. Davis, MD, San Antonio, TX; Nancy Egan, MD, Rockland, ME; Harold F. Farber, MD, Philadelphia, PA; Javier Flores, MD, Miami, FL; Michael H. Gold, MD, Nashville, TN; Terry Jones, MD, Bryan, TX; Cindy Lamerson, MD, Reno, NV; Mark R. Ling, MD, Newnan, GA; Robert W. Loss, MD, Rochester, NY; Michael Maloney, MD, Denver, CO; Bruce Miller, MD, Portland, OR; Eugene Monroe, MD, Milwaukee, WI; David Pariser, MD, Norfolk, VA; Toivo E. Rist, MD, Knoxville, TN; George Russell, MD, Boulder, CO; Thomas J. Russell, MD, Milwaukee, WI; Joel Schlessinger, MD, Omaha, NE; Mary Sheehan, MD, Pittsburgh, PA; Stacy Smith, MD, La Jolla, CA; Dow Stough, MD, Hot Springs, AR; Eduardo Tschen, MD, Albuquerque, NM.
references
1. Buys L. Treatment options for atopic dermatitis. Am Fam Physician. 2007;75:523–528.
2. Leung D, Nicklas R, Li J, et al. Disease management of atopic dermatitis: an updated practice parameter. Ann Allergy Asthma Immunol. 2004;93:S1–S21.
3. Kiyohara C, Tanaka K, Miyake Y. Genetic susceptibility to atopic dermatitis. Allergol Int. 2008;57:39–56.
4. Kay J, Gawkrodger D, Mortimer M, Jaron A. The prevalence of childhood atopic eczema in a general population. J Am Acad Dermatol. 1994;30:35–39.
5. Paller A, Mancini A. Eczematous eruptions in childhood. Hurwitz Clinical Pediatric Dermatology. A Textbook of Skin Disorders of Childhood and Adolescence. Philadelphia: Elsevier-Saunders; 2006:49–64.
6. Mortz C, Lauritsen J, Bindslev-Jensen C, Andersen K. Prevalence of atopic dermatitis, asthma, allergic rhinitis, and hand and contact dermatitis in adolescents. The Odense Adolesence Cohort Study on Atopic Diseases and Dermatitis. Br J Dermatol. 2001;144:523–532.
7. Ozkaya E. Adult-onset atopic dermatitits. J Am Acad Dermatol. 2005;52:579–582.
8. Kapoor R, Menon C, Hoffstad O, et al. The prevalence of atopic triad in children with physician-confirmed atopic dermatitis. J Am Acad Dermatol. 2008;58:68–73.
9. Akdis C, Akdis M, Bieber T, et al. Diagnosis and treatment of atopic dermatitis in children and adults: European Academy of Allergy and Clinical Immunology/American Academy of Allergy, Asthma and Immunology/PRACTALL Consensus Report. Allergy. 2006;61:969–987.
10. Hoare C, Li Wan Po A, Williams H. Systematic review of treatments for atopic dermatitis. Health Technol Assess. 2000;4:1–191.
11. Nilsson E, Henning C, Magnusson J. Topical corticosteroids and Staphylococcus aureus in atopic dermatitis. J Am Acad Dermatol. 1992;27:29–34.
12. Boguniewicz M. Topical treatment of atopic dermatitis. Immunol Allergy Clin N Am. 2004;24:631–644.
13. Williams H. Twice-weekly topical corticosteroid therapy may reduce atopic dermatitis relapses. Arch Dermatol. 2004;140:1151–1152.
14. Green C, Colquitt J, Kirby J, Davidson P, Payne E. Clinical and cost-effectiveness of once-daily versus more frequent use of same potency topical corticosteroids for atopic eczema: a systematic review and economic evaluation. Health Technol Assess. 2004;8:1–120.
15. Lagos B, Maibach H. Frequency of application of topical corticosteroids: an overview. Br J Dermatol. 1998;139: 763–766.
16. Claxton A, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23:1296–1310.
17. Data on file. Medicis Pharmaceutical Corporation, Scottsdale, AZ.
18. VANOS® (fluocinonide) Cream 0.1% [package insert]. Scottsdale, AZ: Medicis Pharmaceutical Corporation; December 2007.
19. Knaysi G, Crikelair G, Cosman B. The rule of nines: its history and accuracy. Plast Reconstr Surg. 1968;41:560–563.
20. Uliasz A, Wong V, Lebwohl M. A single-center, double-blinded study of the atrophogenic effects of fluocinonide cream 0.1% versus clobetasol propionate cream 0.05% in subjects with steroid-responsive dermatoses. Presented at: 31st Hawaii Dermatology Seminar; March 2007; Maui, Hawaii.
21. Gans E, Sadiq I, Stoudemayer T, Stoudemayer M, Kligman A. In-vivo determination of the skin atrophy potential of the super-high-potency topical corticosteroid fluocinonide 0.1% cream compared with clobetasol propionate 0.05% cream and foam and a vehicle. J Drugs Dermatol. 2008;7:10–14.
22. Schlessinger J, Miller B, Gilbert R, Plott T, for the VANOS Study Group. An open-label adrenal suppression study of 0.1% fluocinonide cream in pediatric patients with atopic dermatitis. Arch Dermatol. 2006;142:1568–1572.
23. DiMatteo M. Variations in patients’ adherence to medical recommendations. A quantitative review of 50 years of research. Med Care. 2004;42:200–209.
24. Baldwin H. Tricks for improving compliance with acne therapy. Dermatol Ther. 2006;19:224–236.