Whitney Valins, BS; Clinical Research Fellow, University of Miami Miller School of Medicine, Department of Dermatology and Cutaneous Surgery, Miami, Florida; Sadegh Amini, MD; bSenior Clinical Research Fellow, University of Miami Miller School of Medicine, Department of Dermatology and Cutaneous Surgery, Miami, Florida; Brian Berman, MD, PhD, Professor of Dermatology and Cutaneous Surgery, University of Miami, Miller School of Medicine, Department of Dermatology and Cutaneous Surgery, Miami, Florida
Disclosure: Ms. Valins and Dr. Amini report no relevant conflicts of interest. Dr. Berman serves on the advisory board and speakers bureau of Graceway Pharmaceuticals, LLC.
Abstract
Toll-like receptors are a group of glycoproteins located mostly in cellular membranes, capable of recognizing certain molecules in exogenous microorganisms and initiating immune responses against them through the activation of several intracellular signaling pathways. Toll-like receptors can be stimulated when an inflammatory reaction is needed for the treatment of conditions, such as viral infections or skin cancer, or can be inhibited when a reduction of inflammation is necessary for the treatment of conditions, such as rheumatoid arthritis, systemic lupus erythematosus, and septic shock. In the human skin, keratinocytes and Langerhans cells are known to express these receptors. Skin conditions where Toll-like receptors are known to be upregulated include acne, psoriasis, atopic dermatitis, syphilis, leprosy, Staphylococcus aureus infections, candidiasis, and herpes simplex and varicella zoster infections. Besides imiquimod, which is the most successful and more studied topical Toll-like receptor-modulating agent to date, other topical agents, such as nicotinamide, all-trans retinoic acid, adapalene, zinc, and sodium tosylchloramide, have also been found to exert some of their action through Toll-like receptors. Recent topical agents, including CBT-SL5 and CpG-ODN, are being evaluated for the treatment of inflammatory acne and skin cancer, respectively, and have demonstrated to be effective in the treatment of those conditions. (J Clin Aesthet Dermatol. 2010;3(9):20–29.)
Human Toll-like receptors (TLRs) are a group of glycoproteins that function as surface transmembrane receptors involved in the innate immune response to exogenous pathogenic microorganisms. Prior to the recognition of TLRs, the innate immune system was described as the first proinflammatory line of defense of the body responsible for the recruitment and accumulation of phagocytic cells (i.e., neutrophils and monocytes/macrophages) and the activation of the complement cascade system at micro-organism entry sites. Less than a decade ago, TLRs were found to specifically detect molecular patterns commonly present and conserved among similar groups of microorganisms [pathogen-associated molecular patterns (PAMPs)], such as lipopolysaccharides found on the cell wall of all Gram-negative bacterial species.[1] TLRs then became an important part of the group of receptors that recognize these molecular patterns called “pattern recognition receptors” (PRRs) questioning the nonspecific nature of the innate immune response system. TLRs are found either in the cellular plasma membrane or in intracellular compartments, including the endoplasmic reticulum and endosomes.[2] Ten TLRs have been identified in humans that are capable of recognizing lipids, proteins, and nucleic acids from external sources, such as microorganisms, as well as from internal sources displaying damaged molecular patterns in molecules, such as hyaluronans, heat-shock proteins (HSPs), and fibronectins.[2]
TLR signaling occurs through two distinctive pathways: One dependent on the universal adaptor protein, myeloid differentiation factor 88 (MyD88), and one independent of it. In the MyD88-dependent pathway, there is activation of the nuclear factor-kB (NF-?B) and other kinases, such as c-Jun-NH2-terminal kinase, extracellular signal-regulated kinase (ERK) 1 and 2, and p38 mitogen-activated protein kinases (MAPKs). Upon activation, NF-?B translocates into the nucleus and acts as a transcription factor, while phosphorylation of MAPKs leads to translocation of different transcription factors (i.e., activator protein-1 [AP-1], and Elk-1) into the nucleus. These transcription factors induce the activation of proinflammatory genes coding for inflammatory cytokines.[2–5] MyD88-independent pathways include 1) the alternative activation of NF-?B and MAPKs, bypassing the MyD88 protein complex through downstream activation of TNF receptor-associated factor (TRAF)-6 by TRIF (Toll-IL-1 receptor [TIR] containing adaptor inducing IFN-?); and 2) type-1 interferons (IFNs) gene induction, particularly IFN-? and IFN-? through activation of IFN regulatory factor (IRF)-3 and -7 by TRIF, ultimately inducing the production of inflammatory cytokines, responsible for immune reactions against viruses.
TLRs have also demonstrated, to some extent, control over the adaptive immune system, including T and B cell-mediated and humoral immune responses. TLRs actions related to the adaptive immune responses include the promotion of upregulation of CD40, CD80, and CD86 and the production of interleukin (IL)-12 by dendritic cells (DCs) and monocytes. IL-12 participates in the differentiation of naive T cells into T helper 1 (Th-1) cells, inducing Th-1 cell-mediated immune responses.[2,3,6]
In the human skin, TLRs are expressed in different cell types from the epidermis to the adipose tissue, with great variations in the expression and functionality depending on the cell type. The most important epidermal cells expressing TLRs include keratinocytes, which express TLRs 1-6 and 9, and Langerhans cells (LCs), which can express all TLRs, particularly TLRs 1, 2, 3, 5, 6, and 10. In addition, LCs have been shown to produce IL-10, a Th-2 cytokine, suggesting that LCs may be involved in immunotolerance. Other cells expressing TLRs include resident and trafficking dermal monocytes/macrophages, DCs, T and B lymphocytes, mast cells, endothelial cells, fibroblast, and adipocytes.[2,3,5,7]
TLR activation and inhibition can be influenced by microorganisms as well as by clinicians. As described before, TLRs activate the innate immune system and contribute to further induction of the adaptive immune response to eliminate pathogens upon infection. These pathogens act as TLR agonists eliciting the expression of inflammatory cytokines. However, certain microorganisms, such as Yersinia species and Poxviruses, may act as TLR antagonists, taking advantage of TLR-induced immune responses by evading the host immune response and generating and/or perpetuating infections.[2,5] Clinicians also have access to several agents that function as TLR agonists when induction of immune and inflammatory reactions are needed, for example, for the treatment of viral infections or skin cancer (e.g., TLR 7/8 agonists imiquimod and resiquimod).[5,8] On the other hand, TLRs antagonism (i.e., targeting and blocking TLR stimulation) is a strategy developed for the treatment or prevention of TLR-mediated inflammatory, autoimmune, and infectious conditions, such as rheumatoid arthritis, systemic lupus erythematosus, and septic shock.[9] Finally, mutations and deficiencies in TLR molecules and/or in any of the molecules in the signaling pathways contribute to host susceptibility to develop and perpetuate infections and inflammatory conditions.[2,3,6,10]
SKIN CONDITIONS RELATED TO TLRs
Acne vulgaris. Acne vulgaris is a disorder of the pillosebaceous unit characterized by the excessive production and accumulation of keratin, increased sebum production, and the effect of cutaneous microorganisms, hormones, and inflammatory pathways. Since the 1960s, published data have implicated the anaerobic, Gram-positive bacillus, Propionibacterium acnes, a member of the skin flora colonizing sebaceous follicles, in the etiology of inflammatory acne. P. acnes produces low-molecular weight, serum-independent chemotactic factors that attract neutrophils through the epithelium wall, into the lumen of sebaceous follicles. It also activates the classical and alternative complement pathways, leading to the formation of C5a, an anaphylotoxin that induces the secretion of pro-inflammatory cytokines, including tumor necrosis factor alpha (TNF?), IL-1ß, and IL-8 by monocytes. One of the major components of P. acnes’ cell wall is a very distinctive peptidoglycan, which is an exogenous ligand for TLR-2. Large amounts of TLR-2 have been found to be expressed on perifollicular and peribulbar macrophages in acne lesions. In addition, it has been shown that there is a positive correlation between the severity of acne lesions and the concentration of cells expressing TLR-2. The production of the inflammatory cytokines IL-6, IL-8, and IL-12 is clearly dependent on the interaction of P. acnes and TLR-2. Proinflammatory cytokines secreted by monocytes, IL-8 in particular, are induced through the activation of TLR-2 by P. acnes, leading to the recruitment of neutrophils to the pilosebaceous unit.
In addition to TLR-2, in-vivo and in-vitro studies have demonstrated the expression of TLR-4 in keratinocytes of acne lesions. There is evidence implicating the activation of TLR-4 by P. acnes cell wall lipopolysaccharides in the induction of immune and inflammatory responses in acne.[2,3,5,7,11–14]
Leprosy. Leprosy, also known as Hansen’s disease, is an infectious disease caused by Mycobacterium leprae, an intracellular, Gram-positive bacillus. It is characterized by a broad clinical-histopathological-immunological spectrum. When the host’s immunological response to M. leprae is strong and driven by cell-mediated Th-1 cytokines, such as IFN-?, IL-12, IL-18, and granulocyte-macrophage colony-stimulating factor (GM-CSF), it is considered to be in the tuberculosis form of the disease. On the other hand, a host’s response with Th-2 type cytokines, such as IL-4, and IL-10, that promote a humoral immune response, causes a massive proliferation of bacilli within the tissues and a more disseminated disease characteristic of the lepromatous form of the disease. In-vitro studies have demonstrated that two lipoproteins from M. leprae of 19 and 33 ?Da are capable of mediating cellular activation of monocytes and DCs via TLR-2/1. In a study,[15] incubation of antigen presenting cells (APCs) with 19 ?Da lipoprotein in the presence of Th-1 cytokines resulted in the production of TNF-? by APCs. At the same time, Th-2 cytokines inhibited the production of TNF-? by APCs. In addition, IFN-? induced a strong up-regulation of TLR-1. APCs from skin samples of tuberculoid lesions express TLR-2 and TLR-1 more strongly compared with skin samples of lepromatous lesions. These findings suggest that the activation of TLR-2 and TLR-1 may contribute with the host’s defense against M. leprae.
TLR activation also induces apoptotic pathways. This was demonstrated through activation of TLR-2 expression on the surface of primary human Schwann cells with a synthetic peptide of the 19 ?Da M. leprae lipoprotein, causing an increase in the number of apoptotic Schwann cells. Lipoproteins from M. leprae were able to induce apoptosis on TLR-2-expressing Schwann cells within leprosy lesions and may be responsible for the nerve damage seen in leprosy.[2,3,5,7]
Atopic dermatitis. Atopic dermatitis (AD) is a chronic inflammatory and pruritic skin disease characterized by flares and remissions. There is involvement of a Th-2 cytokine profile of immune response in the skin, and keratinocytes from patients with AD have shown to secrete chemokines attracting eosinophils and Th-2 cells.[5] In patients with AD, flares can be frequently caused by bacterial, viral, and fungal infections, with 90 percent of patients with AD colonized with Staphylococcus aureus in lesional and healthy skin. This increased susceptibility to infections has been explained in part by a reduced amount of several antimicrobial peptides, such as beta-defensins 1 and 2, cathelicidin (LL-37), and dermcidin. Defects in the TLRs or TLR signaling pathways have been identified in AD. Polymorphism in the TLR-2 gene, particularly R753Q, has been shown to be associated with increased susceptibility to and decreased ability to clear S. aureus. In addition, these patients have been associated with severe S. aureus infections. Other polymorphisms, such as in the TLR-9 promoter or in the Toll interactive protein (TOLLIP), an inhibitory adapter protein, have also been associated with AD. [2,3,5]
Psoriasis. Psoriasis is a chronic inflammatory skin disease mediated by T cells, which trigger keratinocytes to hyperproliferate and perpetuate the disease. Psoriasis has been associated with Th-1 and Th-17 cytokine profiles. Several microorganisms have been implicated in the development or exacerbation of the disease; however, contrary to AD (mainly a Th-2-mediated condition), psoriatic plaques are very resistant to superinfections by pathogens, such as S. aureus, partly due to the presence of high levels of antimicrobial peptides in the psoriatic plaques. Keratinocytes from psoriatic plaques express high levels of TLRs 1, 2, 4, 5, and 9, compared with normal skin. Activation of these TLRs has been related to resistance to pathogenic microorganisms. On the other hand, TLR activation has also been implicated in the exacerbation of the disease, as demonstrated by the reaction seen after TLR-7 activation by the topical agonist imiquimod.[16,17] In addition, the antimicrobial peptide cathelicidin (LL-37) has been shown to present self-DNA to activate TLR-9 on plasmacytoid DCs leading to the production of IFN-?, possibly promoting autoimmunity in psoriasis.
The yeast Malassezia furfur, which is related to the development of psoriatic scalp lesions, has been found to upregulate TLR-2 mRNA expression in human keratinocytes. Human b-defensins (HBD)-2 and IL-18 gene expression has been inhibited by anti-TLR-2 antibodies.[2,3,5,7]
Staphylococcus aureus skin infections. S. aureus colonizes the skin and mucous membranes of 20 percent of the human population. It causes infections, such as impetigo, folliculitis, and cellulitis upon crossing the skin barrier. More severe and/or disseminated infections include bacteremia, sepsis, and endocarditis. Several components of the bacteria, such as lipoproteins, peptidoglycans, and lipoteichoic acid, are agonists to TLR-2/6 or TLR-2/2. Signaling pathway through TLR-2 adapter molecule MyD88 and the upregulation of human beta defensin 3 (HBD-3) are major contributors to the immune response against S. aureus. The key molecule seems to be MyD88, which can be activated by other receptors besides TLR-2, including IL-1R. Mice deficient in both MyD88 and IL-1R developed very similar lesions, whereas mice deficient in MyD88 developed larger and more severe lesions than mice deficient in TLR-2. IL-1R seems to be more relevant than TRL-2 in eliciting host immune responses against S. aureus.[2,3,5]
Candida albicans mucocutaneous infections. C. albicans is a dimorphic fungus that causes several mucocutaneous infections, some of them associated with high morbidity and mortality, particularly in immunocompromised patients. Several carbohydrates on the yeast surface, such as glucans, mannans, and chitin, seem to elicit immune responses from the host. Some of these responses have been shown to be generated by recognition and activation of TLRs on keratinocytes. For example, phospholipomannan is recognized by TLR-2, while O-bound mannan is recognized by TLR-4. Activation of TLRs by C. albicans induces the production of proinflammatory cytokines, candidicidal effector molecules, and chemo-attractant molecules to recruit other inflammatory molecules to the site of infection. C. albicans has been shown to manipulate TLRs in order to generate and perpetuate infections in susceptible hosts.[3,5]
Herpesviridae mucocutaneous infections. Host immune reactions to herpes simplex virus (HSV) and varicella-zoster virus, two double-stranded DNA (dsDNA) viruses, are thought to be mediated by several TLRs. Patients with genital HSV infections with TLR-2 abnormalities have been shown to have increased viral shedding and more recurrent episodes than patients without TLR-2 abnormalities. In addition, TLR-3 deficient individuals are more susceptible to HSV encephalitis. In-vitro studies have shown that TLR-9 was able to recognize HSV dsDNA and induce the production of inflammatory cytokines. Varicella-zoster virus has also induced the production of inflammatory cytokines upon activation of TLR-2.3
Cutaneous syphilis. Syphilis is a multistage infection caused by sexual transmission of the spirochete Treponema pallidum, where particular skin lesions, characteristic of each stage, are commonly seen. The interaction of lipopeptides (potent PAMPs) expressed on the surface of T. pallidum with TLR-2 receptors on DCs is thought to be involved in enhancement of the adaptive immune system through T-cell activation and Th-1 cytokine release at the site of antigen exposure. It has been demonstrated that exposure of immature DCs to anti-TLR-2 antibodies led to an inhibition of the T. pallidum lipopeptide-dependent expression of DC surface maturation markers and to the inhibition of DC-dependent T-cell activation.
In addition, the polymers of flagellin, which are key components of the T. pallidum flagellum, function as antigens capable of interacting with DCs as another PAMP. Flagellin polymers bind to TLR-5 and activate NF-?B through MyD88 and IL-1 receptor associated kinase-dependent pathways, leading to production of pro-inflammatory cytokines, such as TNF-?.[5]
Ultraviolet injury. The immune system has been recognized as a fundamental participant in the surveillance against malignant tumors. Recently, it has been found that ultraviolet B (UVB) radiation stimulates the up-regulation and further secretion of HSPs from keratinocyes exposed to UVB. These HSPs are capable of binding to TLRs 2 and 4, and stimulating the Toll/IL-1 pathway in DCs. This signaling induces the downstream production of immunosuppressive cytokines, such as IL-10 and TNF-?. This immunosuppressive effect through TLRs may explain the additional role of UVB in cancer development besides its direct mutagenic effect.[5,18–26]
A summary of several dermatologic conditions in which TLRs upregulation has been demonstrated can be found in Table 1.[2,3,27]
{kind=link}
TLR AGONISTS FOR THE TREATMENT OF SKIN DISEASES
Imiquimod, a member of the family of imidazoquinoline compounds, is the first TLR agonist approved for use in humans.[8] These compounds have potent antiviral and anti-tumor properties. Imiquimod was approved by the United States Food and Drug Administration (FDA) in 1997 for the treatment of genital warts caused by human papillomavirus (HPV). However, it was not until 2002 that the activity of imiquimod and other imidazoquinolines was recognized to be exerted through the activation of the MyD88-dependent TLR-7 signaling pathway, inducing NF-kB and consequently proinflammatory cytokines, including IFN-?, IFN-?, TNF-?, IL-6, IL-8, and IL-12, stimulating the proliferation and maturation of naive (Th-0) lymphocytes, particularly toward the Th-1 phenotype. Furthermore, imiquimod induces LCs to migrate to the lymph nodes, enhancing antigen presentation to T cells. Due to its effective antitumoral activity, imiquimod was also approved in 2004 for the treatment of superficial basal cell carcinoma and actinic keratosis.[28,29] Current off-label indications for imiquimod include Bowen’s disease, lentigo maligna, cutaneous T cell lymphoma, verrucae vulgaris, verrucae plantaris, verrucae plana, and molluscum contagiosum. Recently, generic imiquimod 5% cream became available, and the FDA approved a new concentration, 3.75%, for the treatment of actinic keratosis.[2–5,7,8] Another imidazoquinoline, resiquimod, is 100 times more potent than imiquimod, and is capable of inducing the same cytokine profile through the induction of NF-?B, upon activation of TLRs 7 and 8. Resiquimod has been used mainly for the treatment of actinic keratosis[30] and genital HSV-2 infections.[31,32]
CURRENT TOPICAL AGENTS FOUND TO HAVE TLR ACTIVITY
Nicotinamide. Nicotinamide, an amide derivative of vitamin B3, inhibits cytokine production and leukocyte chemotaxis. It has been used both topically and systemically in several inflammatory disorders including bullous pemphigoid, necrobiosis lipoidica, and dermatitis herpetiformis and has proven to be effective for the treatment of acne.[33] Grange et al[34] studied the molecular mechanisms by which nicotinamide exerted its anti-inflammatory effects on keratinocytes stimulated by P. acnes. Nicotinamide downregulated IL-8 gene expression at transcriptional and post-transcriptional levels and IL-8 protein production in a dose-dependent manner through phosphorylation of the MAPK and TLR-2 dependent I?B degradation, preventing activation of NF?B. In addition, nicotinamide may decrease the half-life of IL-8 mRNA by affecting its stability. Nicotinamide was also found to inhibit ERK and JNK kinase pathways, which have also been implicated in IL-8 synthesis following stimulus from P. acnes.
Poly ADP-ribose polymerase-1 (PARP-1) has also been implicated in NF-?B activation and lipopolysaccharide-induced cytokine expression. Nicotinamide’s effects on IL-8 production may also occur through inhibition of PARP-1. However, further studies are needed to confirm this.[35]
All-trans retinoic acid. All-trans retinoic acid (ATRA) is a derivative of vitamin A with anti-inflammatory properties.[36] In a study by Liu et al,[36] the effect of ATRA on TLR expression in primary human monocytes was studied by measuring mRNA levels and protein expression for TLRs 1–10 and the TLR-2 co-receptor CD14 in cells after 16 hours of ATRA exposure. TLR-2 and CD14 mRNA encoding was decreased by 34 and 65 percent, respectively, along with TLR-1 mRNA encoding (which dimerizes with TLR-2 to take part in ligand recognition). TLR-4 mRNA levels were maintained. ATRA decreased TLR-2 and CD14 protein expression by 41 and 42 percent, respectively, without affecting TLR-1 or TLR-4 protein expression. Due to the lack of retinoic acid receptor (RAR) binding elements present, this regulation of transcription is likely modulated through an alternate signaling pathway.
The effect of ATRA on TLR-induced cytokine release was assessed by treating cells with ATRA for 24 hours followed by stimulation with a TLR-2/1 ligand or a TLR-4 ligand. The TLR 2/1-induced release of IL-12p40, TNF-?, and IL-6 was decreased by 91, 70, and 74 percent, respectively. A weaker effect was seen on TLR-4-induced cytokine release, TLR-2/1-induced-IL-8 release and CD40 expression. In contrast, an effect on TLR-4-induced cytokine release was seen when cells were concurrently treated with TLR-2/1 ligands or TLR-4 ligands and ATRA. IL-12p40 release was reduced by 42 and 44 percent, respectively, TNF-? release was reduced by 56 and 49 percent, respectively, and IL-6 release was decreased by 48 and 37 percent, respectively. Again, no significant effect on IL-8 release or CD40 upregulation was seen. These findings suggest that ATRA directly suppressed TLR-mediated inflammation, most likely by effecting TLR signaling or mRNA stability.
In order to assess ATRA’s role in P. acnes-induced monocyte cytokine release, cells were pretreated with ATRA and then exposed to P. acnes or co-treated with ATRA and P. acnes, followed by measurement of cytokine levels. IL-12p40 and TNF-? were reduced by 53 and 67 percent when cells were pretreated with ATRA while ATRA co-treatment lead to a decrease in the cytokine levels by 37 and 31 percent, respectively. Neither pretreatment nor cotreatment with ATRA affected the levels of IL-6, IL-8, and CD40. These results have helped to facilitate the understanding of an additional mechanism of action of ATRA in the treatment of acne through inhibition of the P. acnes-dependent activation of TLR-2 and TLR-4.
Adapalene. Adapalene is a second-generation topical retinoid derived from naphthoic acid. It selectively binds to nuclear RAR receptors, particularly g and b subtypes found in the epidermis and dermis. In order to confirm results of in-vitro testing[37] implicating adapalene in the dose-depending inhibition of TLR-2 expression by monocytes, Tenaud et al[38] obtained a significant reduction of TLR-2 expression by keratinocytes in a dose-dependent fashion (p=0.026) after 24 hours of incubation of normal human skin samples and acne inflammatory lesions with adapalene at a dose of 10-6M. This was also seen in the suprabasal-layers of the epidermis of acne biopsies at doses of 10-7M (p=0.006) and 10-6M (p=0.012). By decreasing the expression of TLR-2 on keratinocytes in acne, adapalene limits the inflammatory reaction occurring around and in a pilosebaceous follicle. The antagonizing effect of adapalene on TLR-2 seems to be shared by other topical vitamin A derivatives such as ATRA, as discussed above.
Zinc. Zinc is a well-known modulator of inflammatory pathways in acne through various mechanisms including inhibition of polymorphonuclear cell chemotaxis, inhibition of growth of P. acnes, activation of natural killer (NK) cells, and activation of phagocytosis by granulocytes.[39–42] Zinc’s beneficial effect on mild-to-moderate inflammatory acne lesions has been well documented.[43,44] In a study by Jarrousse et al,[45] the anti-inflammatory mechanism of zinc was found to be exerted through modulation of TLR-2 surface expression. Zinc salts significantly decreased TLR-2 expression in normal human epidermal keratinocytes (NHEK) and in skin explants in a dose-dependent manner. This decrease was also seen in keratinocytes treated with zinc after stimulation by P. acnes extracts. Interestingly, zinc salts were not found to affect the level of IL-8 secretion; therefore, IL-8 may not only be controlled by the TLR-2 pathway, but may also be induced by IL-1? and TNF-? production.
Sodium tosylchloramide. Disifin (sodium tosylchloramide) is an efficient disinfectant used in multiple industries. It has the ability to suppress the activity of an array of pathogenic microorganisms, including bacteria, viruses, yeast, and fungi. Its active compound, sodium-N-chloroparatoluenesulphonamide (sodium tosylchloramide) is the first organic chlorine derivative found to have bactericidal effects on the skin[46] through the creation of superoxides by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase.
There is evidence that TLR-4 plays a role in mediating this NADPH-dependent superoxide production that occurs following a microbial infection.[46] This role may include direct interaction of TLR-4 with NADPH oxidase,[47] activation of NF-?B,[48,49] and TLR-4-mediated activation of DCs.[50] Studies to further explore the relationship between Disifin-derived products and TLRs, along with the anti-inflammatory mechanisms of Disifin-derived products may help to create future anti-inflammatory therapies for several inflammatory dermatological conditions.
POTENTIAL TOPICAL AGENTS FOR SKIN CONDITIONS CBT-SL5. As discussed above, it has been suggested that P. acnes induces inflammation through interaction of PAMPs with TLR-2 and TLR-4, leading to activation of a downstream cascade involving NF-?B. Antimicrobial peptides (AMPs) can be induced in the host by pathogenic and by nonpathogenic bacteria colonizing the human body. They play a role in controlling unwanted pathogens such as P. acnes by killing target cells and also controlling inflammation. Enterococcus faecalis SL5, a lactic acid bacteria, produces the AMP CBT-SL5, which has been found to have broad-spectrum antimicrobial activity, including that against P. acnes.[51] The effects of CBT-SL5 on P. acnes-induced inflammation were studied by Lee et al.[52] At concentrations under 100ng/mL, CBT-SL5 was found to significantly reduce the IL-8 mRNA expression at 6 and 12 hours, and IL-8 protein secretion at 24 hours in keratinocytes exposed to P. acnes.
CBT-SL5 was also found to suppress P. acnes-induced translocation of NF-kB into the nucleus, preventing activation of IL-8 gene expression and protein synthesis. This study demonstrated a potential role for CB5-SL5 as a novel therapy in the treatment of inflammatory acne. It has been suggested that it works through interactions with components of P. acnes, acting as a TLR antagonist by preventing the bacteria’s binding to and activation of the TLR cascade; however, its exact site of action in the TLR pathway is unclear and requires further research.
CpG-ODN. Dacarbazine (DTIC) is a standard chemotherapy for the treatment of metastatic malignant melanoma. However, as a mono-therapeutic agent, it has a limited effect in the overall survival of patients. Several studies have examined the role of various adjuvant treatments to enhance the immune response by improving antigen presentation by DCs. TLR agonists represent potential adjuvants for vaccines and cancer immunotherapy. TLR-9 agonists have been shown to induce better cell-mediated, as well as antibody-mediated, immune responses. In addition, TLR-9 agonists are expected to be safer adjuvants, due to their narrow expression profile among TLRs in humans. In a study by Najar et al[53] a chemoimmunotherapy protocol for cutaneous melanoma combining DTIC and topical CpG oligodinucleotide (ODN), a TLR-9 agonist, was evaluated in mice after inoculation with B16 melanoma tumors. The administration of each individual agent, intraperitoneal (IP) DTIC or CpG-ODN (topical or intratumoral), only decreased tumor growth minimally, whereas IP administration of DTIC followed by topical or intratumoral CpG-ODN significantly inhibited tumor growth (p<0.001 and p<0.003, respectively) compared to untreated groups. Of importance, lower tumor volumes and higher survival rates were reported in mice treated with IP DTIC plus topical CpG-ODN than in mice treated with IP DTIC plus intratumoral CpG-ODN. Topical CpG-ODN 250µg was found to be less toxic and more effective than 50µg of subcutaneous CpG-ODN in eliciting immune responses to exogenous antigen. In addition the combination (topical more than intratumoral) induced cellular toxicity and primed cytotoxic-T cells against the tumor. In tumors treated with DTIC and CpG-ODN in combination, a distinct B220+ CD8+ DCE205- CD11c+ population of cells, identical to the phenotype of TLR-9-activated type I IFN-producing plasmacytoid DCs, was observed. This was not seen in tumors treated with DTIC alone. Using cell depletion tests, CD4+ cells and CD8+ cells were confdirmed to be involved in the mechanism of DTIC/topical CpG-ODN in controlling melanoma growth and survival.
Although the results of this study demonstrates the value of topical CpG-ODN as a possible therapy for cutaneous metastatic melanoma, its clinical efficacy in patients with multiple metastases remains to be determined.
SYSTEMIC TLR AGONISTS AND ANTAGONISTS FOR THE TREATMENT OF DERMATOLOGICAL DISEASE
In addition to current and potential topical agents discussed above, several systemic agents targeting TLRs acting as agonists or antagonists are currently being evaluated for the treatment of several dermatological conditions. Discussion of these agents goes beyond the scope of this review, which focuses on topical TLRs. These agents are summarized in Table 2.[8,27,54–57]
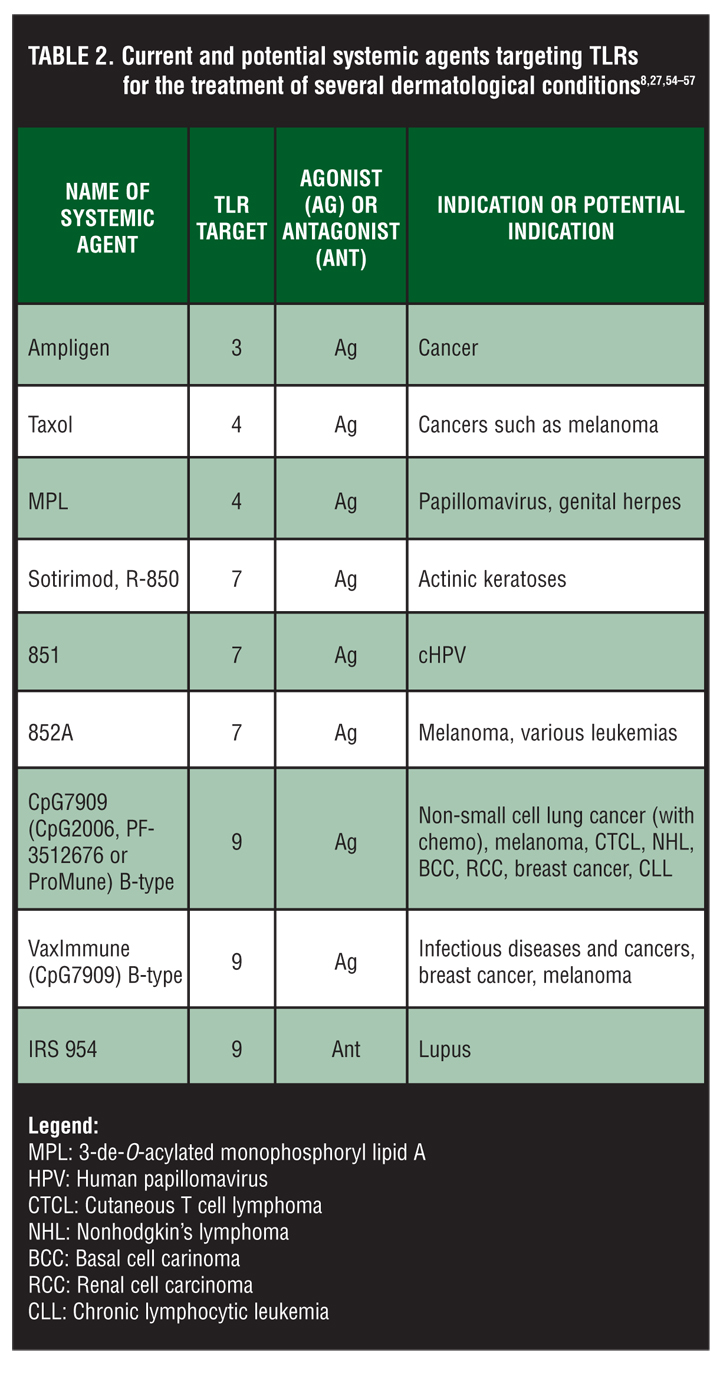{kind=link}
Current studies evaluating agonistic and antagonistic agents targeting TLRs topically or systemically for the treatment of several dermatological conditions are listed in Table 3.[58–65]
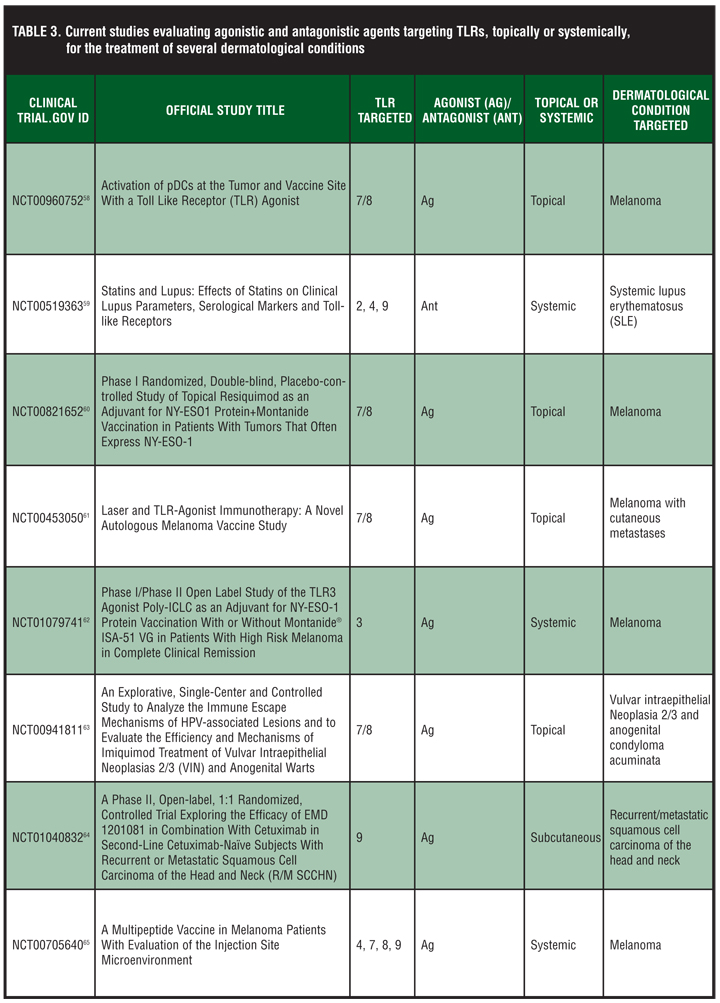{kind=link}
CONCLUSION
Targeting TLRs with agonists (activators) or antagonists (inhibitors) for the treatment of dermatological and other inflammatory conditions represents a growing area of research. Multiple topical therapies, including nicotinamide, ATRA, adapalene, and zinc, have been successfully used for the treatment of acne. Recent research findings have elucidated additional mechanisms of action related to these agents through their important antagonization of the TLR-dependent inflammatory reactions induced by P. acnes in the host. In addition, targeting TLRs with agonists and stimulating immune responses of the host has proven to be a successful adjuvant therapy for the treatment of malignant melanoma. Another example constitutes the successful treatment of superficial basal cell carcinomas, in-situ squamous cell carcinomas (Bowen’s disease), and actinic keratosis by the topical TLR-7 agonist imiquimod. This review has been focused on current and newer topical TLR agonists and antagonists capable of modulating TLRs. These agents represent a growing type of therapy that helps to decrease or stimulate the immune system depending on the nature of the condition being treated. Topical agents modulating TLRs have shown to be effective therapeutic agents for multiple dermatological conditions with the advantage of having an acceptable safety profile and few adverse events seen with the use of systemic therapeutic agents.
References
1. Brightbill HD, Libraty DH, Krutzik SR, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285(5428):732–736.
2. Lai Y, Gallo RL. Toll-like receptors in skin infections and inflammatory diseases. Infect Disord Drug Targets. 2008;8(3):144–155.
3. Miller LS. Toll-like receptors in skin. Adv Dermatol. 2008;24:71–87.
4. Sandor F, Buc M. Toll-like receptors. I. Structure, function and their ligands. Folia Biol (Praha). 2005;51(5):148–157.
5. Kang SS, Kauls LS, Gaspari AA. Toll-like receptors: applications to dermatologic disease. J Am Acad Dermatol. 2006;54(6):951–983.
6. Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469.
7. McInturff JE, Modlin RL, Kim J. The role of toll-like receptors in the pathogenesis and treatment of dermatological disease. J Invest Dermatol. 2005;125(1):1–8.
8. Tomai MA, Miller RL, Lipson KE. Immune response modifiers: imiquimod and future drugs for modulating the immune response. Drug Discov Today Ther Strateg. 2006;3(3):343–352.
9. Tse K, Horner AA. Update on toll-like receptor-directed therapies for human disease. Ann Rheum Dis. 2007;66(Suppl 3):iii77–iii80.
10. Alvarez JI. Inhibition of toll like receptor immune responses by microbial pathogens. Front Biosci. 2005;10:582–587.
11. McInturff JE, Kim J. The role of toll-like receptors in the pathophysiology of acne. Semin Cutan Med Surg. 2005;24(2):73–78.
12. Kim J, Ochoa MT, Krutzik SR, et al. Activation of toll-like receptor 2 in acne triggers inflammatory cytokine responses. J Immunol. 2002;169(3):1535–1541.
13. Marta Guarna M, Coulson R, Rubinchik E. Anti-inflammatory activity of cationic peptides: application to the treatment of acne vulgaris. FEMS Microbiol Lett. 2006;257(1):1–6.
14. Jugeau S, Tenaud I, Knol AC, et al. Induction of toll-like receptors by Propionibacterium acnes. Br J Dermatol. 2005;153(6):1105–1113.
15. Krutzik SR, Ochoa MT, Sieling PA, et al. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat Med. 2003;9(5):525–532.
16. Wu JK, Siller G, Strutton G. Psoriasis induced by topical imiquimod. Australas J Dermatol. 2004;45(1):47–50.
17. Gilliet M, Conrad C, Geiges M, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol. 2004;140(12):1490–1495.
18. Noonan FP, DeFabo EC, Kripke ML. Suppression of contact hypersensitivity by UV radiation and its relationship to UV- induced suppression of tumor immunity. Photochem Photobiol. 1981;34:683–689.
19. Rivas JM, Ullrich SE. Systemic suppression of delayed-type hypersensitivity by supernatants from UV-irradiated keratinocytes. An essential role for keratinocyte-derived IL-10. J Immunol. 1992;149:3865–3871.
20. Yoshikawa T, Kurimoto I, Streilein JW. Tumor necrosis factor alpha mediates ultraviolet light enhanced expression of contact hypersensitivity. Immunology. 1992;76:254–271.
21. Kock A, Schwarz T, Kirnbauer R, et al. Human keratinocytes are a source for tumor necrosis factor alpha: evidence for synthesis and release upon stimulation with endotoxin for ultraviolet light. J Exp Med. 1990;172:1609–1614.
22. Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1998;22:631–677.
23. Vabulas RM, Ahmad-Nejad P, da Costa C, et al. Endocytosed HSP60 uses toll-like receptor 2 (TLR2) and TLR4 to activate the toll/ interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–339.
24. Vabulas RM, Braedel S, Hilf N, et al. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll- like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–20853.
25. Vabulas RM, Ahmed-Nejad P, Ghose S, et al. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112.
26. Caramalho I, Lopes-Calcalho T, Oster D, et al. Regulatory T cells selectively express Toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411.
27. Romagne F. Current and future drugs targeting one class of innate immunity receptors: the Toll-like receptors. Drug Discov Today. 2007;12(1-2):80–87.
28. Berman B, Amini S, Valins W, Block S. Pharmacotherapy of actinic keratosis. Expert Opin Pharmacother. 2009;10(18):3015–3031.
29. Berman B, Viera M, Amini S, Valins W. Immune response modulators in the treatment of skin cancer. In: Rigel et al, ed. Cancer of the Skin. 2nd ed. Chapter 44. Philadelphia, Pa: Elsevier, Inc. In press.
30. Szeimies RM, Bichel J, Ortonne JP, et al. A phase II dose-ranging study of topical resiquimod to treat actinic keratosis. Br J Dermatol. 2008;159(1):205–210.
31. Fife KH, Meng TC, Ferris DG, Liu P. Effect of resiquimod 0.01% gel on lesion healing and viral shedding when applied to genital herpes lesions. Antimicrob Agents Chemother. 2008;52(2):477–482.
32. Mark KE, Corey L, Meng TC, et al. Topical resiquimod 0.01% gel decreases herpes simplex virus type 2 genital shedding: a randomized, controlled trial. J Infect Dis. 2007;195(9): 1324–1331.
33. Shalita AR, Smith JG, Parish LC, et al. Topical nicotinamide compared with clindamycin gel in the treatment of inflammatory acne vulgaris. Int J Dermatol. 1995;34(6): 434–437.
34. Grange PA, Raingeaud J, Calvez V, et al. Nicotinamide inhibits Propionibacterium acnes-induced IL-8 production in keratinocytes through the NF-kappaB and MAPK pathways. J Dermatol Sci. 2009;56(2):106–112.
35. Kolb H, Burkart V. Nicotinamide in type 1 diabetes. Mechanism of action revisited. Diabetes Care. 1999;22(Suppl 2):B16–B20.
36. Liu PT, Krutzik SR, Kim J, et al. Cutting edge: all-trans retinoic acid down-regulates TLR2 expression and function. J Immunol. 2005;174(5):2467–2470.
37. Vega B, Jonard A, Michel S. Regulation of human monocyte Toll-like receptor 2 (TLR2) expression by adapalene (abstract). J Eur Acad Dermatol Venereol. 2002: 16:123–124.
38. Tenaud I, Khammari A, Dreno B. In-vitro modulation of TLR-2, CD1d and IL-10 by adapalene on normal human skin and acne inflammatory lesions. Exp Dermatol. 2007;16(6): 500–506.
39. Dreno B, Trossaert M, Boiteau HL et al. Zinc salts effects on granulocyte zinc concentration and chemotaxis in acne patients. Acta Derm Venereol. 1992;72:250–252.
40. Strauss JS, Stranieri AM. Acne treatment with topical erythromycin and zinc: effect of Propionibacterium acnes and free fatty acid composition. J Amer Acad Dermatol. 1984;11:86–89.
41. Dreno B, Foulc P, Reynaud A, et al. Effect of zinc gluconate on propionibacterium acnes resistance to erythromycin in patients with inflammatory acne: in-vitro and in-vivo study. Eur J Dermatol. 2005;15(3):152–155.
42. Chvapil M, Stankova L, Zukoski CT et al. Inhibition of some functions of polymorphonuclear leukocytes by in-vitro zinc. J Lab Clin Med. 1977;89:135–146.
43. Dreno B, Amblard P, Agache P et al. Low doses of zinc gluconate for inflammatory acne. Acta Derm Venereol. 1989;69:541–543.
44. Dreno B, Moyse D, Alirezai M, et al. Acne Research and Study Group. Multicenter randomized comparative double-blind controlled clinical trial of the safety and efficacy of zinc gluconate versus minocycline hydrochloride in the treatment of inflammatory acne vulgaris. Dermatology. 2001;203(2): 135–140.
45. Jarrousse V, Castex-Rizzi N, Khammari A, et al. Zinc salts inhibit in vitro Toll-like receptor 2 surface expression by keratinocytes. Eur J Dermatol. 2007;17(6):492–496.
46. Ofodile ON. Disifin (sodium tosylchloramide) and Toll-like receptors (TLRs): evolving importance in health and diseases. J Ind Microbiol Biotechnol. 2007;34(12):751–762.
47. Park HS, Jung HY, Park EY, et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 173:3589–3593.
48. Beutler B. TLR4: central component of the sole mammalian LPS sensor. Curr Opin Immunol. 2000;12:20–26.
49. Underhill DM, Ozinsky A. Toll-like receptors: key mediators of microbe detection. Curr Opin Immunol. 2002;14:103–110.
50. Vulcano M, Dusi S, Lissandrini D, et al. Toll receptor-mediated regulation of NADPH oxidase in human dendritic cells. J Immunol. 173:5749–5756.
51. Van Reenen C, Van Zyl WH, Dicks LM. Expression of the immunity protein of plantaricin 423, produced by Lactobacillus plantarum 423, and analysis of the plasmid encoding the bacteriocin. Appl Environ Microbiol. 2006;72: 7644–7651.
52. Lee YJ, Choi HJ, Kang TW, et al. CBT-SL5, a bacteriocin from Enterococcus faecalis, suppresses the expression of interleukin-8 induced by Propionibacterium acnes in cultured human keratinocytes. J Microbiol Biotechnol. 2008;18(7): 1308–1316.
53. Najar HM, Dutz JP. Topical CpG enhances the response of murine malignant melanoma to dacarbazine. J Invest Dermatol. 2008;128(9):2204–2210.
54. Meyer T, Stockfleth E. Clinical investigations of Toll-like receptor agonists. Expert Opin Investig Drugs. 2008;17(7): 1051–1065.
55. Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117(5):1184–1194.
56. Jasani B, Navabi H, Adams M. Ampligen: a potential toll-like 3 receptor adjuvant for immunotherapy of cancer. Vaccine. 2009;27(25-26):3401–3404.
57. Ospelt C, Gay S. TLRs and chronic inflammation. Int J Biochem Cell Biol. 2010;42(4):495–505.
58. Activation of pDCs at the Tumor and Vaccine Site With a Toll Like Receptor (TLR) Agonist. ClinicalTrials.gov Identifier: NCT00960752. http://clinicaltrials.gov/ct2/show/ NCT00960752. Accessed on May 17, 2010.
59. Statins and Lupus: Effects of Statins on Clinical Lupus Parameters, Serological Markers and Toll-like Receptors. ClinicalTrials.gov Identifier: NCT00519363. http:// clinicaltrials.gov/ct2/show/NCT00519363. Accessed on May 17, 2010.
60. Phase I randomized, double-blind, placebo-controlled study of topical resiquimod as an adjuvant for NY-ESO1 protein+montanide vaccination in patients with tumors that often express NY-ESO-1. ClinicalTrials.gov Identifier: NCT00821652. http://clinicaltrials.gov/
ct2/show/NCT00821652. Accessed on May 17, 2010.
61. Laser and TLR-agonist immunotherapy: a novel autologous melanoma vaccine study. ClinicalTrials.gov Identifier: NCT00453050. http://clinicaltrials.gov/ct2/show/
NCT00453050. Accessed on May 17, 2010.
62. Phase I/Phase II open label study of the TLR3 agonist poly-ICLC as an adjuvant for NY-ESO-1 protein vaccination with or without montanide® ISA-51 VG in patients with high risk melanoma in complete clinical remission. ClinicalTrials.gov Identifier: NCT01079741. http://clinicaltrials.gov/ct2/show/
NCT01079741. Accessed on May 17, 2010.
63. An explorative, single-center and controlled study to analyze the immune escape mechanisms of HPV-associated lesions and to evaluate the efficiency and mechanisms of imiquimod treatment of vulvar intraepithelial neoplasias 2/3 (VIN) and anogenital warts. ClinicalTrials.gov Identifier: NCT00941811. http://clinicaltrials.gov/ct2/show/
NCT00941811. Accessed on May 17, 2010.
64. A Phase II, open-label, 1:1 randomized, controlled trial exploring the efficacy of EMD 1201081 in combination with cetuximab in second-line cetuximab-naive subjects with recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN). ClinicalTrials.gov Identifier: NCT01040832. http://clinicaltrials.gov/ct2/show
/NCT01040832. Accessed on May 17, 2010.
65. A multipeptide vaccine in melanoma patients with evaluation of the injection site microenvironment. ClinicalTrials.gov Identifier: NCT00705640. http://clinicaltrials.gov/ct2/show/ NCT00705640. Accessed on May 17, 2010.