 J Clin Aesthet Dermatol. 2020;13(4):40–42
J Clin Aesthet Dermatol. 2020;13(4):40–42
by Lucia Gallo, MD; Matteo Megna, MD; Bianca Festa, MD; Pio Stellato, MD; Rosita Di Pinto, MD; Gabriella Fabbrocini, MD; and Maria Ferrillo, MD
Drs. Gallo, Megna, Festa, Fabbrocini, and Ferrillo are with the Section of Dermatology in the Department of Clinical Medicine and Surgery at the University of Naples Federico II in Napoli NA, Italy. Drs. Stellato and di Pinto are with the Section of Pediatrics in the Department of Translational Medical Science at the University of Naples Federico II in Napoli NA, Italy.
FUNDING: No funding was provided for this study.
DISCLOSURES: The authors have no conflicts of interest relevant to the content of this article.
ABSTRACT: Background. Rowell syndrome is a rare disease characterized by a combination of lupus erythematosus (LE) and erythema multiforme (EM)-like lesions and a characteristic immunologic pattern, including speckled pattern of antinuclear antibody (ANA), positive anti-Ro/SSA or anti-La/SSB, and positive rheumatoid factor (RF). This disease was first described in 1963, and to date, about 95 cases of EM-like lesions associated with LE have been described in the literature, with the majority of cases being reported in adult female patients. Here, we describe the case of a 17-year-old male patient exhibiting both systemic LE and EM lesions together with ANA and anti-Ro/SSA positivity who was successfully treated with systemic corticosteroids and hydroxychloroquine.
KEYWORDS: Rowell syndrome, erythema multiforme, lupus erythematosus
Rowell syndrome (RS) is a rare disease characterized by a combination of lupus erythematosus (LE) and erythema multiforme (EM)-like lesions and a characteristic immunologic pattern, including speckled pattern of antinuclear antibody (ANA), positive anti-Ro/SSA or anti-La/SSB, and positive rheumatoid factor (RF). The association between LE and EM was first described in 1922, but the definitive recognition of their association as a separate disease entity, named Rowell disease, was described in 1963.1,2 To date, about 95 cases of EM-like lesions associated with LE have been described in literature, with the majority of the cases being reported in female patients (female to male ratio 8:1).3 We describe the case of a 17-year-old male patient exhibiting systemic LE and EM-like lesions together with ANA positivity.
Case Report
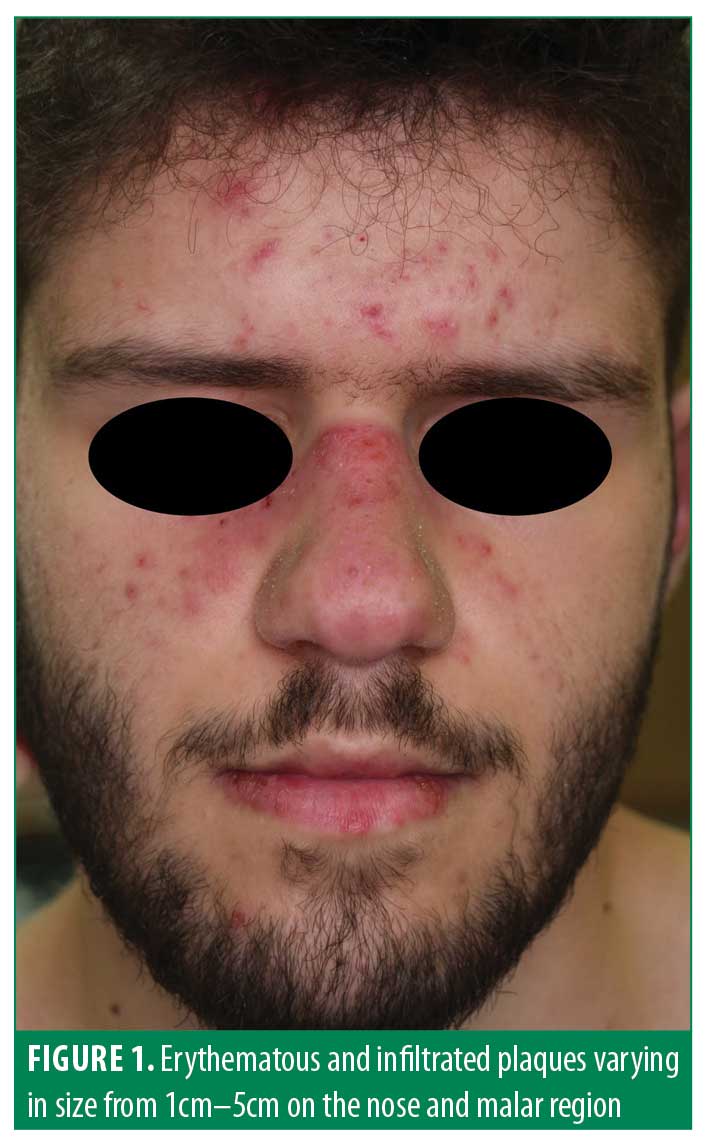
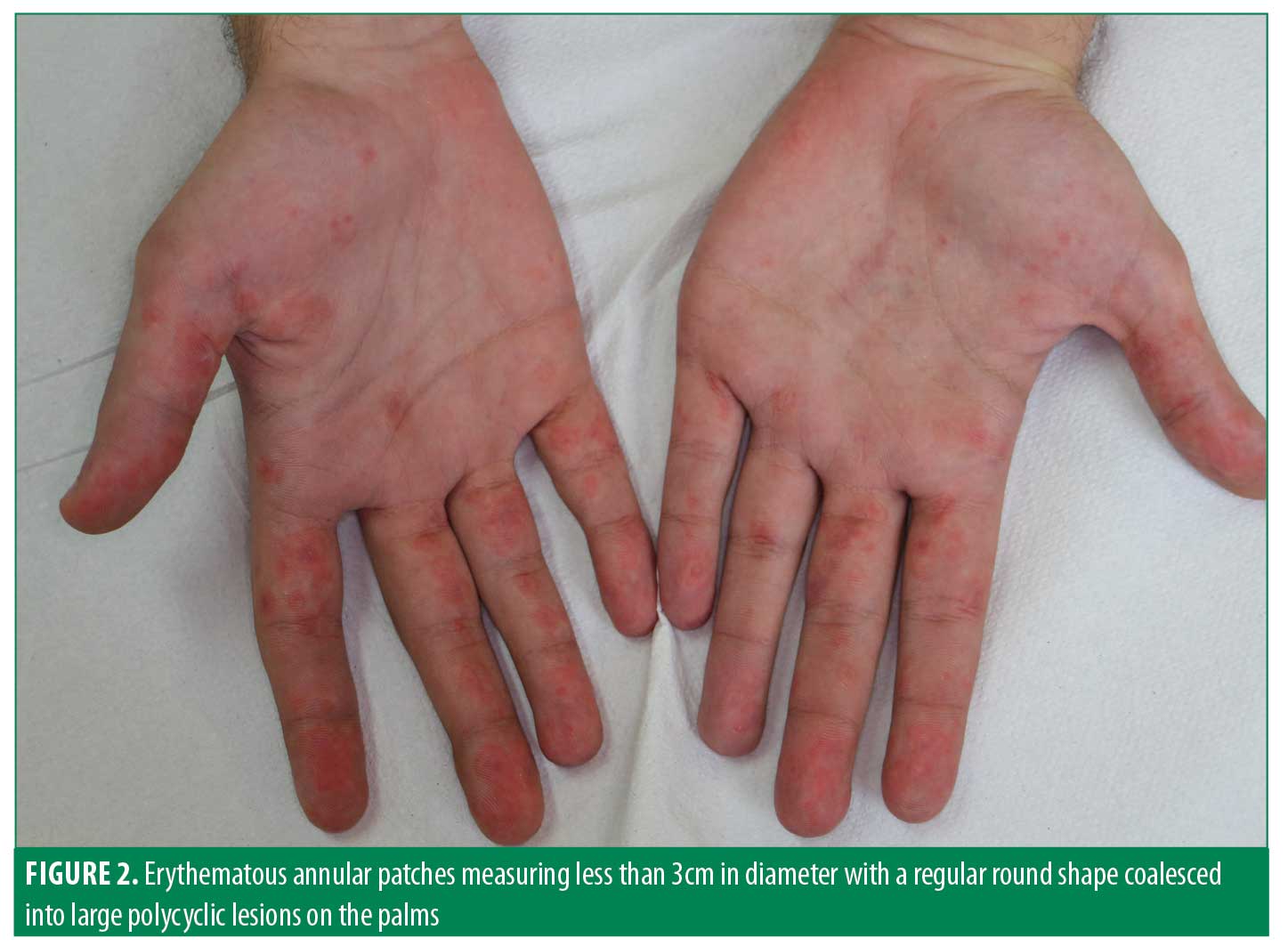
A 17-year-old male patient presented to our clinic with a one-month history of facial rash, photosensitivity, and fever. The patient also reported severe pain in his knees and shoulders. The patient had a history of perisplenic lymphadenopathy and hepatomegaly in the three months prior to visiting our clinic . He was not taking any medication. Clinical examination showed asymptomatic erythematous and infiltrated plaques varying in size from 1cm to 5cm on the nose and malar region which resembled classical cutaneous LE lesions (Figure 1). However, erythematous, annular patches measuring less than 3cm in diameter with a regular round shape which coalesced into large polycyclic lesions were also observed on the palms, lips, and mouth, suggesting typical EM (Figure 2). The patient related that these EM-like lesions developed almost two months before admission to our clinic. Clinical examination did not reveal any ocular, oral, and/or genital lesions. After an collecting the patient’s medical history, possible iatrogenic and/or infectious triggers were not identified. Laboratory investigations showed a decreased white blood cell count (3310/mm3 n.v. 4,800–10,800) and an increase in erythrocyte sedimentation rate (45mm per hour, n.v. <15mm per hour), and fibrinogen (447mg/dl, n.v. 160–350). ANA were positive (18.5, n.v. 0–1.00) and ENA-profile showed the presence of anti-Ro/SSA (297.5 UA/mL, n.v. <10); anti-Sm (130.4 UA/mL, n.v. <10); anti-RNP (135.8 UA/mL, n.v. <10); and anti-dsDNA (600 IU/ml, n.v. <30). Renal and cardiac function were normal.
 A 3-mm skin biopsy taken from an erythematous plaque on the patient’s nose showed orthokeratotic stratum corneum, focal interface changes, and peri-appendageal mononuclear cell infiltrate consistent with a diagnosis of cutaneous LE. Based on medical history and clinical, histological, and laboratory findings, a diagnosis of systemic LE was proposed. Indeed, the patient met the diagnostic criteria for systemic LE by the presence of skin rash, arthralgia, leukopenia, and positive antibodies, according to SLICC classification criteria.4 However, a 5-mm punch biopsy of a targetoid lesion located on the palmar area showed the presence of keratinocyte necrosis and perivascular lymphocytic infiltrate with superficial edema in the dermis.
A 3-mm skin biopsy taken from an erythematous plaque on the patient’s nose showed orthokeratotic stratum corneum, focal interface changes, and peri-appendageal mononuclear cell infiltrate consistent with a diagnosis of cutaneous LE. Based on medical history and clinical, histological, and laboratory findings, a diagnosis of systemic LE was proposed. Indeed, the patient met the diagnostic criteria for systemic LE by the presence of skin rash, arthralgia, leukopenia, and positive antibodies, according to SLICC classification criteria.4 However, a 5-mm punch biopsy of a targetoid lesion located on the palmar area showed the presence of keratinocyte necrosis and perivascular lymphocytic infiltrate with superficial edema in the dermis.
These features together with clinical aspect supported the diagnosis of EM. Therefore, given the presence of both LE and EM lesions, as well as the positivity of ANA and anti-Ro/SSA, a final diagnosis of RS was determined. The patient was started on prednisone 1mg/kg daily and hydroxychloroquine 200mg twice daily, which resulted in gradual improvement of skin manifestations and resolved related symptoms at a two-month follow-up. After tapering systemic corticosteroids, the patient was still taking hydroxychloroquine 200mg twice daily at the six-month follow-up and no recurrence of the disease had been observed.
Discussion
RS is a disease characterized by both LE and EM-like lesions in subjects with a characteristic immunologic pattern. The disease usually appears in young adults. The median age reported is 32 (range 9–87); however, pediatric or elderly cases have also been described. RS appears to be more prevalent in female patients than male patients (female to male ratio is 8:1) and only eight patients were under 18.3 The diagnosis of LE usually precedes the appearance of EM-like lesions.3 However, the detailed chronological history of the lesions is not reported in all the described RS cases. In our patient, the appearance of LE and EM lesions was almost simultaneous.
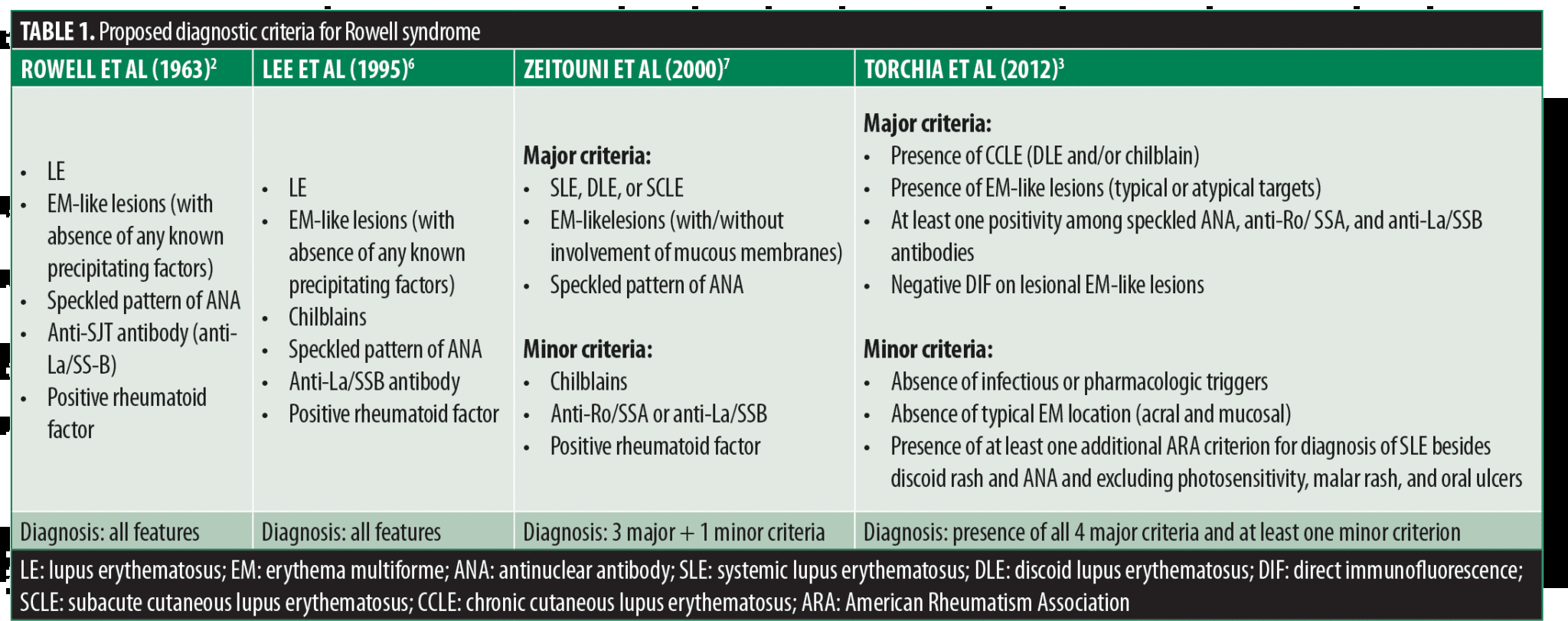
Classic EM is often caused by bacterial and viral infections (e.g., mycoplasma pneumonia, herpes simplex virus), drugs (e.g., antibiotics, anticonvulsants, nonsteroidal anti-inflammatory drugs, tuberculostatic drugs), or malignancies.5 On the other hand, EM is not associated with any specific serological abnormalities commonly seen in autoimmune disease.5 EM cases associated with LE lesions where an EM trigger factor is missing are considered an RS diagnostic criterion.2,6 In 1963, Rowell et al2 first described diagnostic criteria for RS: presence of discoid lupus erythematosus (DLE) and EM-like lesions, positive RF, speckled ANA, and a saline extract of human tissue (anti-SJT), now known as similar to anti-Ro/SSA positivity. In 1995, Lee et al6 observed the presence of chilblains in cases of RS. In 2000, Zeitouni et al7 divided the diagnostic criteria into major and minor. The major criteria included LE (acute, subacute, or systemic), EM-like lesions, and antinuclear antibodies positivity. The minor criteria were the presence of chilblains, and presence of anti-Ro/SSA or anti-La/SSB antibodies, and rheumatoid factor. All major criteria and at least one minor criterion are necessary for an RS diagnosis.
However, diagnostic criteria for RS are still debated. In 2011, Torchia et al3 proposed new diagnostic criteria and defined RS as an independent chronic cutaneous lupus erythematosus (CLE) subtype (Table 1). Antiga et al8 suggested that LE with EM-like lesions represents a subset of subacute cutaneous lupus erythematosus (SCLE) with targetoid lesions rather than a distinct entity, and in a minority of cases, only a coincidental association, like between CLE and lichen planus or psoriasis. This conclusion was based on a review of the literature by Antiga et al that revealed the majority of EM cases included in RS were more consistent with the annular-polycyclic variant of SCLE than with EM.
Histologic findings as diagnostic criteria for RS are controversial. The role of necrotic keratinocytes is still debated. Torchia et al3 focused only on negative direct immunofluorescence of EM lesions, stating that there were no significant histological differences between EM and CLE lesions. Indeed, the presence of necrotic keratinocytes is not specific for EM because it might also be found in 54 percent of SCLE lesions.
Our patients met the diagnostic criteria for RS defined by both Zeitouni and Torchia.3,7 Moreover, our patient did not show any identifiable precipitating factor for EM. Interestingly, our patient was a young man (<18 years old) despite the majority of RS cases being reported in middle-aged female patients.10
The prognosis and treatment are similar to those of systemic lupus erythematosus or DLE that occur alone. In most cases of RS, prednisone combined with azathioprine, antimalarials (e.g., chloroquine, hydroxychloroquine), dapsone, or cyclosporine were frequently reported.11,12
Conclusion
RS is a rare entity which has been regarded as only a coincidence, an overlapping syndrome, a variant of CLE, or a true “ghost syndrome.”13 To date, there is not a consensus classification of RS to avoid misdiagnosis of this syndrome. We report this case to highlight the necessity to suspect RS in all patients, even younger male patients, with LE and EM-like lesions even without evidence of a precipitating factor. RS might still be an underdiagnosed and underestimated disease. Further cases must be signaled so that progress in RS pathophysiology knowledge and diagnostic criteria can be achieved.
References
- Scholtz M. Lupus erythematosus acutus disseminates haemorrhagicus. Arch Dermatol Syphilol. 1922;6:466.
- Rowell NR, Beck JS, Anderson JR. Lupus Erythematosus and Erythema multiforme-like lesions. Arch Dermatol. 1963;88:176–180.
- Torchia D, Romanelli P, Kerdel FA. Erythema multiforme and Stevens-Johnson syndrome/toxic epidermal necrolysis associated with lupus erythematosus. J Am Acad Dermatol.
2012;67:417–21. - Sag E, Tartaglione A, Batu E, et al. Performance of the new SLICC classification criteria in childhood systemic lupus erythematosus: a multicentre study. Clin Exp Rheumatol. 2014;32:440–444.
- Branisteanu DE, Ianosi SL, Dimitriu A, et al. Drug-induced Rowell syndrome, a rare and difficult to manage disease: a case report. Exp Ther Med. 2018;15(1):785–788.
- Lee S, Schloss E, Kowichi J. Rowell’s syndrome: a case report with subacute cutaneous lupus erythematosus and erythema multiforme. Can J Dermatol. 1995;7:807-10.
- Zeitouni NC, Funaro D, Cloutier RA, et al. Redefining Rowell’s syndrome. Br J Dermatol. 2000;142: 343–346.
- Antiga E, Caproni M, Bonciani D, et al. The last word on the so-called ‘Rowell’s syndrome’? Lupus. 2012;21:577–585.
- Herrero C, Bielsa I, Font J et al. Subacute cutaneous lupus erythematosus: clinicopathologic findings in thirteen cases. J Am Acad Dermatol. 1988;19: 1057–1062.
- Madke B, Khopkar U. Rowell’s syndrome in an Indian male and review of the literature. Indian Dermatol Online J. 2015;6:S12–S16.
- Lee A, Batra P, Furer V, et al. Rowell syndrome (systemic lupus erythematosus + erythema multiforme. Dermatol Online J. 2009;15:1.
- Bhat RY, Varma C, Bhatt S, Balachandran C. Rowell syndrome. Indian Dermatol Online J. 2014;5: S33–S35.
- Bonciolini V, Antiga E, Caproni M, Fabbri P. Rowell syndrome: does it exist? Clin Exp Dermatol. 2014;39:58.