LindaSusan Marcus, MD; Private Practice, Wyckoff, New Jersey; Director–Chair, Department of Dermatology, Valley Hospital, Ridgewood, New Jersey;Neal Carlin, BS, LindaSusan Marcus, MD, Wyckoff, New Jersey; Robert Carlin, MA, LindaSusan Marcus, MD, Wyckoff, New Jersey.
Disclosure: The authors report no relevant conflicts of interest.
Abstract
This case report highlights a rare, virulent, aggressive, poorly differentiated, epithelioid fibrosarcoma with numerous rhabdoid cells on the tibial aspect of an 84-year-old woman. Biopsy of the lesions were reviewed at College of Physicians and Surgeons of Columbia University Department of Dermatopathology (New York, New York) and in consultation with the Soft Tissue Pathology Division of the Surgical Pathology Department at Columbia University as well as the Department of Pathology at the Massachusetts General Hospital Division of Bone and Soft Tissue Pathology. The management of this patient was difficult especially considering her mental status. She had an excellent response to radiation therapy. It is important to be innovative in treatment and to use judgment when assessing unusual entities.
(J Clin Aesthet Dermatol. 2010;3(12):50–53.)
An 84-year-old woman initially presented to her internist with lesions on her leg that had been consistently developing and were noted when the patient suffered a cerebral vascular accident in May 2009. She had been taking anticoagulants since 1997. The lesions bled easily; therefore, they were assumed to be hemorrhagic bulli, but they did not dissipate and new lesions appeared. In August 2009, the patient was seen in dermatology.
The patient has garbled speech and dementia secondary to two prior cerebrovascular accidents (CVAs) that have been documented with multiple infarcts on magetic resonance imaging (MRI) of the brain. She has glaucoma, macular degeneration, and is legally blind. She had colitis in 1998. Her past skin history includes vasculitis, phlebitis, urticaria, and eczema and is significant for a biopsy-proven seborrheic keratosis, which was removed in 1995 from the tibial area in the same location of the presenting lesions.
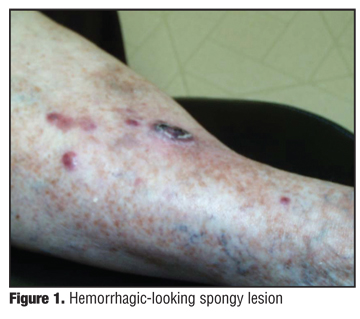
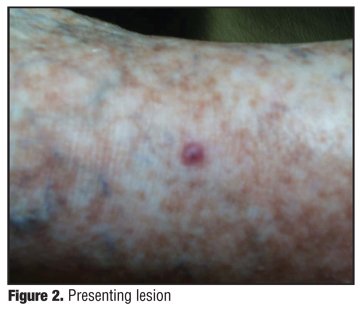
The patient takes Coumadin (warfarin) 2.4 four days a week, Lexapro (escitalopram oxalate) 10mg, Synthroid (levothyroxine) 0.45mg, digoxin 0.25mg, Excelon Patch (rivastigmine transdermal system) 0.5mg, Entocort (budesonide) 3mg, Lipitor (atorvastatin) 20mg, Seroquel (quetiapine) 12.5 at bedtime, and Klor-con (potassium chloride) 10mg twice daily. On physical examination, there were 10 3mm to 2.5cm nontender, hemorrhagic, purple, compressible but firm, soft lesions on the anterior right tibia (Figure 1 and Figure 2). No inguinal lymphadenopathy was present. The rest of the physical examination was unremarkable. The computed tomography (CT) scan of the chest and MRI of the leg were negative. An MRI of the brain shows evidence of the earlier CVAs with no acute changes.
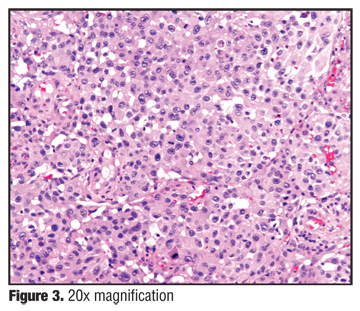
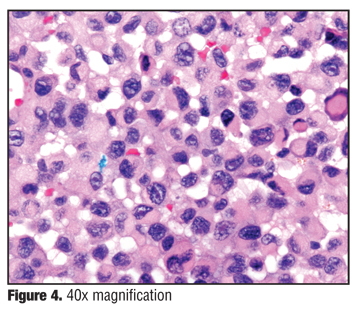
Histology, which was read at Columbia Presbyterian Department of Dermatopathology in consultation with the Soft Tissue Pathology Division of the Surgical Pathology Department at Columbia as well as the Department of Pathology at the Massachusetts General Hospital Division of Bone and Soft Tissue Pathology, had features of an undifferentiated malignant neoplasm. There is a dense cellular infiltrate composed of atypical cells with vacuolated cytoplasm and nucleoplasm and atypical nuclear chromatin patterns. There is suggestion of goblet cells and many vessels with hemorrhage. There are tumor cells in the dermis, some of which have a rhabdoid phenotype. An extensive proliferation of blood vessels was also seen (Figure 3 and Figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The tumor showed a pseudo-sinusoidal pattern reminiscent of a malignant hemangioendothelioma or angiosarcoma; however, vascular immunostains did not support this interpretation. Immunostaining was negative for pan-cytokeratin and for epithelial membrane antigen, militating against a carcinoma. Staining for S-100, HMB-45, and Melan-A, as well as for Bcl-2, was negative, militating against melanoma, including conjunctival melanoma. Staining was negative in the large tumor cells for CD34 and FLi1 as well as for CD31 and factor VIII, although these showed focally a slight blush attributable to the discohesive nature of this tumor and the presence of some serum in artifactual spaces. Tests for estrogen and progesterone were negative. Immunostaining was positive for vimentin and CD10, but this is not helpful in determining the specific cell lineage of the tumor.
Wide excision was not feasible considering the extent of the lesions including the satellite areas. Although chemotherapy including two courses of doxorubicin 50mg/M every two weeks during radiation therapy was considered, the patient’s other problems and general condition obviated this approach.
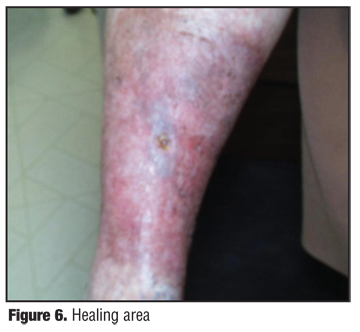
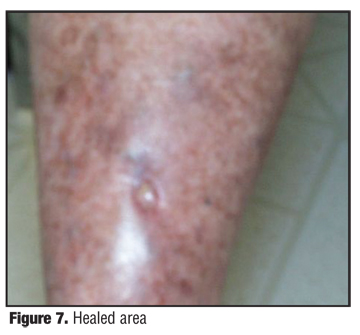
The patient initially received radiation therapy 5300 centigray at 180 centigray per fraction in 35 fractions over a period of two months, with a combination of lateral 6MV photons and phos 6MeV electron, all based on CT scan for three-dimensional planning with custom wedges, bolus, and fields. She had a cone down to the primary lesion of 900 centigray. The cone down was administered utilizing phos 9MV electrons in custom bolus output. A larger field was treated initially based on the position of the clinical lesions present on the skin and the location of the satellite nodules that had been previously biopsied. The patient tolerated the therapy well and responded excellently to this treatment, which is similar to the therapy for Kaposi’s sarcoma (Figure 4). The patient experienced regression of the smaller satellite lesions as well as the primary central lesion that involuted as an ulcer (Figure 5). She developed a radiation dermatitis that subsided with topical steroid cream (Figure 6 and Figure 7).
{kind=link}
{kind=link}
{kind=link}
Discussion
Clinically, the patient’s lesions resembled a Kaposi-like vascular neoplasm; however, the histology did not support this diagnosis. Many negative studies, including mucicarmine stain, did not reveal epithelial mucin and immunostaining for pan-cytokeratin and for epithelial membrane antigen militated against carcinoma. Staining for S-100, HMB-45, Melan-A, and Bcl-2 militated against melanoma including conjunctival melanoma. Staining was also negative in the large tumor cells for CD31, FLi1, CD34, CK7, CK20, CDX2, TTF-1, GCDFP-15, CD3, CD20, CD30, CD43, CD45, CD1a, CD138, CD163, MUM1, and factor VIII. EMA, carbonic anhydrase IX (CA 9), CD56, estrogen, progesterone, desmin, and smooth muscle actin were negative. WT1 was present only in cytoplasm and was therefore considered negative. With these studies, a lymphoid neoplasm was also eliminated. INI1 staining was not lost. Immunostaining was positive for vimentin and CD10, but this is not helpful in determining the specific cell lineage of the tumor. Angiosarcoma was also eliminated due to negative endothelial markers. This lesion may represent a poorly differentiated, epitheloid fibrosarcoma with numerous rhabdoid cells that is felt to be aggressive in nature. It may also represent a composite and must, in itself, be considered an entity of its own.
There have been sporadic reports in the literature of unusual tumors that are a diagnostic dilemma and become a treatment problem.[1] Although many lesions have been confused with osteosarcoma, chondrosarcoma, and angiosarcoma, primary malignant rhabdoid tumors have been reported with the expression of vimentin in the tumor cells and no other positive results, similar to this patient.[2,3] Aggressive malignant rhabdoid tumors presenting as hemangiomas of unknown etiology have been seen in neonates.[4] It was thought that epithelioid sarcomas and malignant rhabdoid tumors were two distinct entities; however, they have similar electron microscopic and immunohistochemical features and may represent the same or related entities. The rhabdoid lesions have a poorer prognosis.[5] Epithelioid sarcomas with positive histochemical markers, including vimentin and CD34, were reported indicating these mesenchymal neoplasms are capable of partial epithelial transformation since no single immunomarker could be found that would categorize different tumors into definitive subtypes.[6] This phenotypic versus entity controversy has existed for decades.[7]
Conclusion
It was interesting to monitor the progression of this rare malignant neoplasm of the leg. New entities are constantly evolving, especially in an aging population. This case is perhaps the eighth reported case of a primary malignant rhabdoid tumor. Physicians must be aware of variations of entities and modify therapy accordingly considering the patient and the diagnosis. Sporadic reports of rare lesions exist, and clinicians must have an open mind when confronted with lesions that do not seem to fit a known pattern. It is important to remember there will always be new and rare entities to treat. Keeping an open mind with a rational clinical judgment can help lead to a treatment plan that is efficacious, feasible, and offers an appropriate cosmetic result.
References
1. Suarez-Viteleia D, Izquierdo-Gracia F, Alonso-Orcajo N. Epithelioid and rhabdoid rhabdomyosarcoma in an adult patient: a diagnostic pitfall. Virchows Archiv. 2004;445(3): 323–325.
2. Folpe AL, McKennney JK, Brigde JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults. Report of four cases of hyalinizing matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma or angiosarcoma. Am J Surg Pathol. 2002;26:1175–1183.
3. Dabbs DJ, Park HK. Malignant rhabdoid skin tumor: an uncommon primary skin neoplasm. Ultrastructural and immunohistochemical analysis. J Cutan Pathol. 2006;15(2): 109–115.
4. Albregts AE, Hebert AA, Aboul-Nasr RA, Raney B. Malignant rhabdoid tumor presenting as a hemangioma. Pediatr Dermatol. 2008;13(6):468–471.
5. Perrone T, Swanson PE, Twiggs L, Ulbright TM, Dehner LP. Malignant rhabdoid tumor of the vulva. Is distinction from epithelioid sarcoma possible? A pathologic and immunohistochemical study. Am J Surg Pathol. 1989;13: 848–858.
6. Laskin W, Miettinen M. Epithelioid sarcoma: new insights based on an extended immunohistochemical analysis. Arch Pathol Lab Med. 2003;127(9):1161–1168.
7. Ognino S, Ro JY, Redline RW. Malignant rhabdoid tumor: a phenotype? An entity? A controversy revisited. Adv Anat Pathol. 2000;7:181–190.