aMarisa Wolff, DO; aChristopher Mancuso, DO; bKaran Lal, BS; cDamian DiCostanzo, MD; a,dCharles Gropper, MD
aSt. Barnabas Hospital Dermatology Residency, Bronx, New York
bNew York College of Osteopathic Medicine-NYIT, Old Westbury, New York
cPathology Associate Pc, Dermatopathology, Portchester, New York
dMount Sinai School of Medicine Manhattan, New York
Disclosure: The authors report no relevant conflicts of interest.
Abstract
Dermatomyositis is a myopathic or amyopathic autoimmune connective tissue disease that presents with classic dermatologic findings ranging from: poikilodermatous photosensitivity (shawl sign), eyelid edema and violaceous-pigmentation (heliotrope sign), lichenoid eruptions on the knuckles and elbows (Gottron’s sign), periungual telangiectasias, and ragged cuticles (Samitz sign). Up to 30 percent of adult-onset cases of dermatomyositis may represent a paraneoplastic syndrome warranting a thorough work-up for malignancy. The authors present a case report of paraneoplastic dermatomyositis associated with triple negative, BRCA-1 positive, invasive intraductal carcinoma of the breast, whose myopathic and cuteanous symptoms were recalcitrant to high-dose corticosteroid therapy. Herein, the authors describe the first reported case of the use of an injectable adrenocorticotropic hormone agonist gel in a patient with myopathic paraneoplastic disease that achieved clinical resolution of both myopathic and cutaneous symptoms, but subseuqently developed significant hyperpigmentation of her face suspected to be secondary to a chemotherapeutic-induced pigmentary change which was augmented by adrenocorticotropic hormone therapy. J Clin Aesthet Dermatol. 2017;10(1):57–62
A 55-year-old Hispsanic woman presented to the dermatology clinic complaining of an “itchy rash” and “swollen face” that developed gradually over two months. These pruritic lesions were initially pink and underwent gradual purple darkening with associated swelling of the skin on her face. The skin eruption progressed to involve her chest, proximal upper extremities, and upper back. She had been seen in the emergency room three times over the past two months and had failed empiric therapy with oral prednisone and diphenhydramine for a suspected allergic reaction. Her history was limited to mild seasonal allergies. Review of systems was significant for two weeks of worsening upper extremity weakness and myalgia as well as fatigue and weight loss over the last two months. She denied fever or any recent infectious illness and current medications included only those provided by the emergency room. Upon review of electronic medical record, it was found that on her third visit to the emergency room she had additionally complained of a breast mass and had recently visited a breast surgeon with a biopsy revealing invasive intraductal carcinoma of the breast. The patient was not aware of these results at the time of the initial dermatology visit.
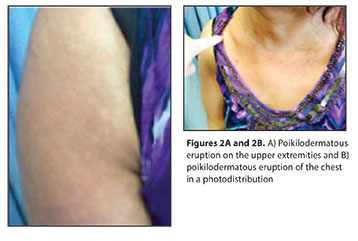
On physical examination, there was striking generalized facial edema with mauve-pink to violaceous patches on eyelids, forehead, and medial malar cheeks extending into her nasolabial folds bilaterally (Figure 1). There was a photodistributed and poikilodermatous eruption of confluent red violaceous patches involving arms, chest, and upper back (Figure 2). Inspection of nails showed ragged cuticles. Clinical differential diagnosis included paraneoplastic dermatomyositis, contact dermatitis, photoallergic drug eruption, airborne contact dermatitis, systemic lupus erythematosus, cutaneous T-cell lymphoma, lichen planus actinicus, rosacea, and granuloma faciale. The intensity of facial edema allowed for the differential to also include scleromyxedema, primary systemic amyloidosis, Hansen’s disease, and sarcoidosis.
{kind=link}
{kind=link}
A punch biopsy was obtained from her upper back revealing perivascular lymphocytes and interface dermatitis with vacuolar alteration at the dermal epidermal junction and scattered necrotic keratinocytes (Figure 3). These findings were consistent with a diagnosis of dermatomyositis, and clinical findings supported the paraneoplastic subtype.
{kind=link}
Laboratory testing was significant for an elevated creatine kinase of 838 IU/L, and positive anti-nuclear antibodies (ANA) with a titer of 1:160. Rheumatologic immunoassays were negative for anti-Jo, anti-sm, anti-U1RNP, anti-dsDNA, anti-histone, anti-cardiolipin, and anti-scl70 antibodies. Anti-P155 antibody testing was not available, but the patient did have an elevated CA-125 tumor marker as well as a positive BRACA 1 gene mutation. Oncologic workup revealed the diagnosis of triple negative, stage IV invasive intraductal carcinoma of the breast with nodal metastasis.
Therapy initially began with medium and high potency topical steroids with poor control of cutaneous disease. She was next started on high-dose oral prednisone, which moderately reduced facial edema and myositis-related discomfort; however, symptoms quickly flared during taper. During the taper oncology began her chemotherapeutic regimen of gemcitabine and carboplatin. Myositis symptoms continued to progress and she developed inability to dress herself or perform other activities of daily living. Due to the worsening of myositis as well as recalcitrant cutaneous disease (in conjunction with rheumatology and oncology), she was started on a trial of subcutaneous injections of Acthar® Gel (Mallinckrodt Pharmaceuticals)—a highly purified analogue of adrenocorticotropic hormone (ACTH) in gelatin at a dose of 80 U/mL biweekly for 12 weeks duration.
Following 12 weeks of ACTH-gel injections, in addition to chemotherapy, her creatine kinase levels quickly normalized, occurring in parallel to the resolution of her proximal upper extremity weakness and myalgias. Cutaneous lesions also improved with resolved facial edema and a resolution of her heliotrope sign as well as shawl sign over her 12 weeks of therapy (Figure 3). While her poikilodermatous skin changes resolved, she paradoxically developed a marked photodistributed facial hyperpigmentation (Figure 4) as well as melanonychia of her fingernails following therapy.
{kind=link}
The authors’ patient has completed seven cycles of chemotherapy with no new foci of metabolic tumor activity. The tumor has undergone a slight decrease in size; however, there remains to be no change in metabolic activity on PET scan. During the two months following discontinuation of ACTH therapy, her dermatologic and muscle symptoms have remained quiescent, and her creatine kinase level has remained normalized. The authors continue to monitor her closely for reoccurrence of DM symptoms and are currently only treating her hyperpigmentation with regular sun protection.
Discussion
Dermatomyositis (DM) is an autoimmune connective tissue disease that can present with or without associated inflammatory myopathy. The overall incidence of disease is approximately 1/100,000 and is 2 to 3 times more common in women.[1] The majority of cases are idiopathic in nature; however, the paraneoplastic subtype is seen in up to 30 percent of cases of adult-onset DM.1 Age of onset tends to be bimodal; however, juvenile cases are not associated with an increased risk of malignancy.[2]
Paraneoplastic DM was first described in 1916 by Sterz who noted a tendency toward female predominance, particularly after their fifth decade of life. The most common associated underlying neoplasm is ovarian cancer; however, 20 percent of cases of paraneoplastic DM have been associated with a primary breast carcinoma.1 The high frequency of associated breast carcinoma is not surprising, as breast cancer is common with a lifetime incidence in the United States of 1 in 8 women.1 Other frequently associated malignancies include colon, lung, prostate, pancreatic, and gastric carcinomas as well as non-Hodgkin’s lymphoma.
The pathophysiology behind the development of DM is yet to be elucidated; however, it is suspected to result from an underlying alteration in cellular and humoral immunity. This alteration has been associated with a response to drugs, infectious agents, or malignancy in genetically predisposed individuals.[2] Paraneoplastic DM is believed to be the result of interactions between several tumor-derived biologic mediators, such as hormones, peptides, antibodies, cytotoxic lymphocytes, autocrine, and paracrine mediators.[3]
Paraneoplastic symptoms are caused by a malignancy, but are not directly related to invasion by the tumor or its metastases.[4] Specific etiology remains poorly understood, but the possibility of autoantibody cross reactivity has been suggested. This theory has been supported by the finding of myositis-specific autoantigen, histadyl tRNA synthetase (HRS/Jo-1) expressed at higher levels in myositis muscles and regenerating muscle cells in addition to carcinoma of the lung, breast, and liver.[4] These findings support the possibility that myositis symptoms may result from cross-reactivity between auto-antigens against cancer cells as well as those of regenerating muscle cells. Another theory suggests that tumor-associated antigens may be produced by neoplastic inflammatory cells and their production may be increased in the setting of autoimmune disease.[5] In addition to anti-Jo-1, autoantibodies to transcriptional intermediary factor 1 gamma (155kDa) (anti-p155 Ab) have also been found in adult onset DM associated with internal malignancy.
The link between rheumatic disease and paraneoplastic phenomenon has been extensively investigated. It is known that systemic inflammatory rheumatic diseases may increase the risk for the development of malignancies, particularly lymphoproliferative disorders.[3] This increased risk applies to connective tissue diseases, such has rheumatoid arthritis, systemic lupus erythematous, scleroderma, and dermatomyositis. Sustained inflammatory activity seems to be the primary risk factor for malignancies in these autoimmune diseases.[3] In cases where malignancy follows rheumatologic disease, immunosuppressive drugs and biological agents have also been suggested to play a role in carcinogenesis.[3]
Clinical presentation of both DM and paraneoplastic DM are similar in that a classic, photodistributed pink-violet poikilodermatous eruption develops involving the eyelids (heliotrope sign), and the v-of the chest and upper back (shawl sign). Heliotrope sign often begins with swelling around the eyes spreading into the cheeks followed by pink violaceous discoloration. Lesions of DM can often be distinguished from lupus due to the former being more violaceous in color while the latter being primarily red in color. Well-marginated, violaceous, and lichenoid papules may also develop over knuckles (Gottron’s papules), and elbows or knees (Gottron’s sign). Examination of the hands may show nail-fold telangectasias and ragged cuticles (Samitz sign). Associated symptoms may or may not include proximal weakness, muscle pain and atrophy. It is important to note that both DM and the paraneoplastic variant can be amyopathic. The juvenile DM variant may also be amyopathic and may present with secondary findings of calcinosis cutis.
Serum studies in paraneoplastic DM are analogous to classic DM, with elevated muscle enzymes, such as creatine kinase, adolase, and lactate dehydrogenase. These muscle enzymes serve as markers of disease activity and are released due to autoimmune damage and inflammation of muscle tissues. The utility of routine laboratory testing for autoantibodies in DM is not well established with serum ANA positivity in less than 30 percent of cases.[2],[4] Immunologic assays for anti-Jo-1, anti-Mi-2, and anti-U1-RNP antibodies are specific to DM, but are often negative due to low sensitivity. There are no definitive paraneoplastic markers; however, anti-p155 antibodies are more commonly found in cases of cancer-associated DM with myositis with specificity of 89 percent, sensitivity of 70 percent, and negative predictive value of 93 percent.[4]
Diagnosis of DM is made most commonly by Bohan and Peter criteria, which include typical rash of DM, symmetrical muscle weakness, muscle biopsy evidence of myositis, elevated serum muscle enzymes, and characteristic electromyography (EMG) findings. Definitive diagnosis of DM requires a skin or muscle biopsy in the setting of clinical disease. Histological findings include thinning of the epidermis, hydropic degeneration of basal layer, basement membrane thickening, papillary dermal edema, and a perivascular and periadnexal lymphocytic infiltrate within dermis. These histological findings are difficult to distinguish from cutaneous lupus and may require a clinical and serologic correlation.
Adult patients diagnosed with DM should be evaluated closely for personal and family history of cancer and undergo age and history-appropriate malignancy screening. As previously stated, up to 30 percent of all cases of adult DM are paraneoplastic. Most underlying cancers will occur within two years of DM diagnosis, so patients should be screened for at least 3 to 5 years following the initial diagnosis.[1],[5] Laboratory screening tests may include serology for prostate specific antigen (PSA), CA-125, or fecal occult blood studies for prostate, ovarian, and colon cancer screening, respectively. Clinicians may consider ordering a computed tomography (CT) of the thorax, abdomen, and pelvis in men and women over 40 years old. Women over the age of 40 are recommended to undergo comprehensive examination with an ultrasound of the pelvic region followed by CT, mammography, and an magnetic resonance imaging (MRI) of the breast.[7] An ultrasound of the testes is recommended in any adult man presenting with DM under the age of 50 years. If primary screening is negative, it is judicious to repeat screening after 3 to 6 months and screen every six months up until 4 to 5 years. It is important to keep in mind that a cutaneous eruption and myopathy may relapse in the setting of recurrent malignancy, and these patients should continue to be followed closely for dermatologic signs of reoccurrence.[1]
Treatment of underlying malignancy will usually result in symptom resolution in cases of paraneoplastic DM. Recurrence of tumor is often associated with a recurrence of DM. Sunlight avoidance and sunscreen may help alleviate skin manifestations. Topical therapies with corticosteroids or calcineurin inhibitor may provide some relief of symptoms in select patients. In the setting of severe cutaneous and/or myositis symptoms, various immunosuppressive medications such as systemic corticosteroids, methotrexate, or hydroxychloroquine have been utilized with variable results. While high-dose systemic corticosteroids are often first-line treatment in classic DM, paraneoplastic cases tend to be less responsive to systemic steroid therapy.[4] In addition to chemotherapeutics and radiation therapy for underlying malignancy, systemic therapy for dermatomyositis symptoms with intravenous immunoglobulin (IVIG), rituximab, methotrexate, and hydroxycholoroquine have been utilized with variable success.
ACTH gelatin injection was approved by the United States Food and Drug Administration (FDA) in 2010 for the treatment of DM in the setting of exacerbation, recalcitrant disease, or maintenance therapy. The mechanism of action is believed to mimic ACTH and induce steroidogenesis, resulting in anti-inflammatory and immunomodulatory effects with disease clearance. This therapy has shown to be effective in patients refractory to steroids and is also FDA approved for the treatment of lupus, psoriatic arthritis, rheumatoid arthritis (RA), juvenile RA, multiple sclerosis, serum sickness, and respiratory sarcoidosis.
There are no current case reports of ACTH therapy being utilized in paraneoplastic dermatomyositis. While the current patient’s cutaneous and myopathic symptoms responded drastically to a 12-week treatment course, she also developed significant hyperpigmentation of her face (Figure 4 and Figure 5). Acthar Gel is a long-acting formulation of full sequence ACTH that includes other proopiomelanocortin peptides, which are precursors to both ACTH as well as melanocyte-stimulating hormone.[6] Cutaneous hyperpigmentation is a well-known side effect of chemotherapy, particularly seen with alkylating agents (i.e., carboplatin) and antimetabolites (i.e., gemcitabine); however, it is possible that this pigmentary change was further augmented by the administration of an ACTH agonist through the inadvertent delivery of melanocyte-stimulating hormone precursors.
{kind=link}
Conclusion
DM presenting in an adult should raise clinical suspicion of an underlying contributing malignancy. The development of DM as a paraneoplastic syndrome may follow the identification of an underlying malignancy or may present months to years before clinical manifestation of the malignancy. Since most cancers occur within two years of DM diagnosis, current recommendations suggest that patients should be screened for at least 3 to 5 years following the initial diagnosis. As dermatologists, it is our role to identify these heralding signs of underlying malignancy and provide our patients with the opportunity for earliest possible detection and therapeutic management. It is the authors’ hope that this case report and review of paraneoplastic DM will enhance recognition and screening for this variant and suggest ACTH-gel injections as another therapeutic option for the subset of patients with recalcitrant myopathic disease.
References
1. Sandhu N, Zakaria S, Degnium A, Boughey J. Dermatomyositis presenting as a paraneoplastic syndrome due to underlying breast cancer. BMJ Case Rep. 2011 Feb 2.
2. Bolognia JL, Jorizzo JL, Rapini RP. Dermatomyositis. In: Dermatology. 3rd ed. China: Elsevier; 2012: 632–640.
3. Szekanecz Z, Szekanecz E, Bako G, Shoenfeld Y. Malignancies in autoimmune rheumatic diseases—a mini review. Gerontology. 2011;57: 3–10.
4. Wada C, Hua C, Carney M. Paraneoplastic syndrome in Hawaii: a case of dermatomyosisitis associated with endometrial cancer. Hawaii J Med Public Health. 2014;73(4): 112–114.
5. Velez A, Howard M. Diagnosis and treatment of cutaneous paraneoplastic disorders. Dermatol Ther. 2010;23(1):662–675.
6. Levine T. Treating refractory dermatomyositis or polymyositis with adrenocorticotropic hormone gel: a retrospective case series. Drug Des Devel Ther. 2012;6:133–139.
7. Titulaer MJ, Soffietti R, Dalmau J, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS Task Force. Eur J Neurol. 2011;18(1):19–e3.
8. Yasar S, Gurleyik G, Sabuncuoglu Y, et al. Paget’s disease of the breast in a patient with amyopathic dermatomyositis. Case Reports in Medicine. 2012;(5):515691.