Jorge Garcia-Zuazaga, MD, MS, Director, Mohs Micrographic Surgery, Department of Dermatology, University Hospitals Westlake Medical Center; Malcolm S. Ke, MD, Division of Dermatology, University of California Los Angeles; Marlene Willen, MD, Chair, Department of Dermatology, MetroHealth Medical, Cleveland, Ohio
Abstract
Cysts are entities encountered frequently in dermatological clinics. Various types of cysts have been described and include trichilemmal cysts, epidermoid cysts, steatocystomas, and the myriad of developmental cysts (branchial cleft cyst, thyroglossal duct cysts, bronchogenic cysts). Moreover, not all lesions that appear clinically as cystic structures are, in fact, cysts. Increased awareness of these mimickers and a systematic approach to the evaluation of these cases is essential. The authors report seven cases, over the course of six years, presenting to their dermatology department, all of which were originally clinically diagnosed as “cysts” and referred to the authors for management. In this article, the authors review seven cyst mimickers and describe important aspects of these diagnoses to increase awareness of the importance of a preoperative biopsy and evaluation. It is important to have a thorough understanding of the wide differential diagnosis of cutaneous nodules and to consider other causes of lesions that appear to be cysts, particularly in the anatomical locations described. (J Clin Aesthetic Dermatol. 2009;2(10):28–33.)
Cysts and cyst-like structures are frequently seen in dermatological practices. Histologically, a true cyst contains a closed cavity surrounded by a wall of true epidermis. The cyst cavity is filled with keratinaceous or fatty material arranged in layers.[1] In contrast, cyst-like structures, although lacking these histological markers, may be clinically identical to true cysts because of similar anatomical location and morphological appearance. It is important to differentiate between these cysts mimickers because true cysts are usually treated by simple surgical excision; whereas, other cyst-like masses may require further diagnostic workup and a more aggressive surgical approach.[2]
It is necessary to have a clear understanding of the various cyst mimickers and to consider these entities when encountering patients with cutaneous nodules.
Over the past six years, many interesting cases initially diagnosed as epidermal inclusion cysts were referred for excision to the authors’ dermatology clinic. Many had clinically changed in morphology, size, or color by the time they reached the authors’ clinic, while others remained benign-appearing. These patients were referred to the authors’ department for evaluation of a suspected cyst. This article presents seven cases of cyst mimickers: hidradenoma, cutaneous B-cell lymphoma, epithelioid sarcoma, Merkel cell carcinoma, metastatic adenocarcinoma of the lung, granular cell tumor, and cutaneous meningioma to increase awareness that other lesions can mimic cysts.
Case Series
Case 1. A 58-year-old Caucasian man was referred for evaluation of an asymptomatic growth over his left chest, which was present for two and half months. He reported “clear yellow fluid” upon aspiration by a nurse practitioner the week prior. Physical examination revealed a 2x2cm, firm, nontender, subcutaneous nodule over the left lateral chest. No lymphadenopathy was appreciated. Initial biopsy revealed a nodular hidradenoma. Subsequent complete excision was performed.
Case 2. A 41-year-old Caucasian man was referred for excision of an asymptomatic enlarging nodule over the frontal scalp for the past six months. His past medical history was significant for irritable bowel syndrome. Physical examination revealed a 2.5×2.5cm, erythematous, firm tumor with overlying alopecia and telangiectasia. Excision of the tumor revealed a cutaneous B-cell lymphoma. He underwent a course of rituximab followed by local radiation and is currently in remission.
Case 3. A 37-year-old Caucasian woman was referred for evaluation of an asymptomatic nodule over her left posterior parietal scalp for the past 22 years. She had a family history of melanoma. Physical examination revealed a 1x1cm nontender, indurated nodule. Excisional biopsy revealed a high-grade, malignant, spindled and epithelioid sarcoma with osteosarcomatous component. She underwent wide local excision followed by local radiation treatment and has been in remission for the past three years.
Case 4. A 31-year-old Caucasian man was referred for excision of an asymptomatic, rapidly growing nodule over the chest. Physical examination revealed a 5x5cm indurated tumor with overlying telangiectasia and, centrally, a 2cm protuberant nodule. Skin biopsy revealed Merkel cell carcinoma. The patient underwent primary resection and a positive axillary lymph node dissection followed by chemotherapy. He had been in remission for almost two years, but recently returned with meningeal, bone, and mediastinal metastases.
Case 5. A 41-year-old Caucasian woman was referred for evaluation of a painful left occipital scalp nodule for the past four months. On initial presentation, she was diagnosed with an infected epidermoid cyst and given systemic antibiotics. A few weeks later, the patient complained of generalized myalgias and pain radiating from her “cyst” to her right jaw and right temple. She also noticed several new lumps appearing on both sides of her chest wall and abdomen. Physical examination revealed a 2cm, indurated, erythematous, tender nodule over the left occipital scalp and multiple subcutaneous nodules over the bilateral axillae and abdomen. Biopsy of the left occipital scalp and left flank revealed cutaneous adenocarcinoma metastases. Further evaluation over the following weeks revealed Stage 4 non-small cell adenocarcinoma of the lung with metastases to the brain and adrenal gland. She died two weeks later.
Case 6. A 64-year-old Caucasian man presented with a several-month history of a subcutaneous lesion overlying the right parietal scalp. Physical examination showed a 3x4cm, smooth, slightly tender, hard mass. His neurological examination was unremarkable. A computed tomography (CT) scan demonstrated the lesion without a bony defect or intracranial connection. Pathological diagnosis was cutaneous meningioma. The patient refused further excision of the lesion.
Case 7. A 47-year-old African-American woman presented to our clinic for evaluation of a lump on her left anterior thigh for the past year. She denied trauma or drainage. This lesion was asymptomatic and had been growing for the past month. Examination showed a well-circumscribed, painless, subcutaneous nodule measuring 1.3×1.8cm. Biopsy showed granular cell tumor. Subsequent complete excision was performed with no sequelae.
Discussion
The differential diagnosis of a subcutaneous nodule is very broad and often provides the dermatologist with a complex diagnostic challenge (Table 1).[1–3] In addition to the myriad of cysts (epidermoid, trichilemmal, mucous, ganglion, embryologic, steatocystomas), subcutaneous masses may also arise from any cell type (mesenchymal, neural, adipose, adnexal), presenting as benign or their malignant counterparts.[3–5]
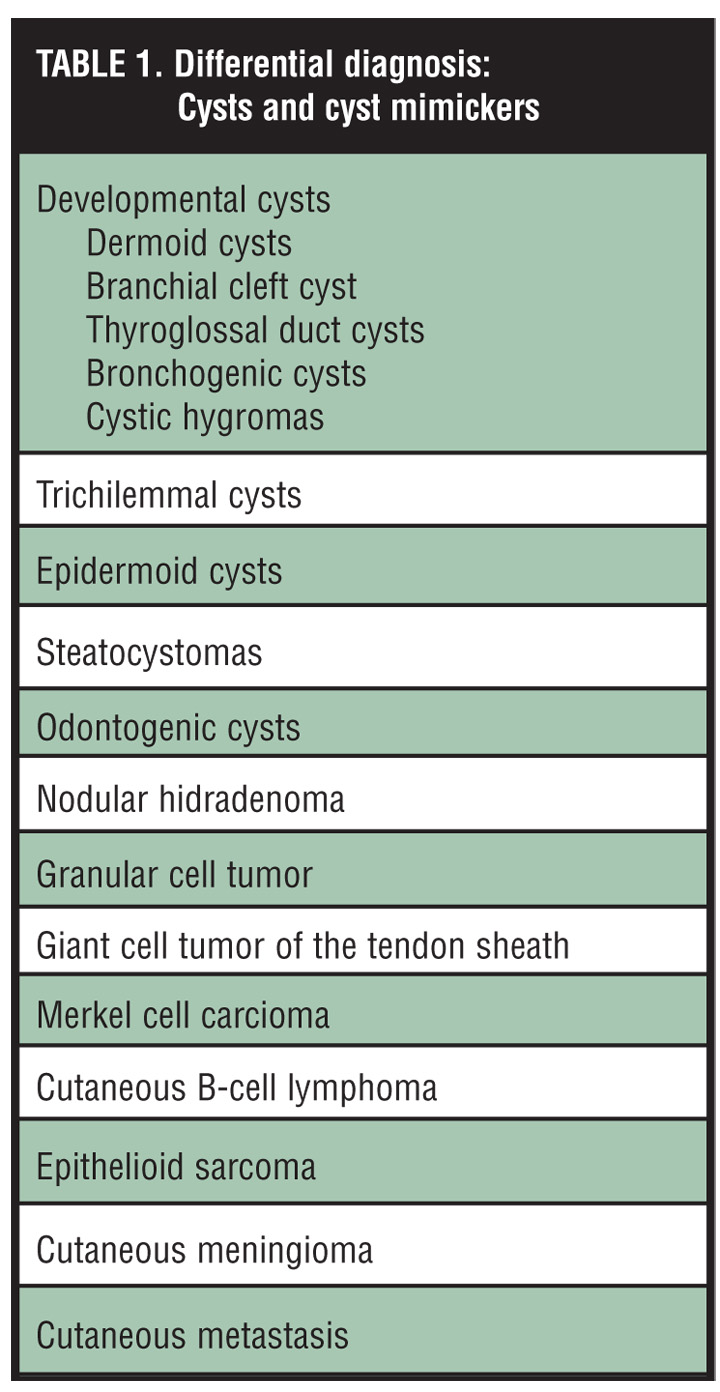{kind=link}
Furthermore, developmental cysts (usually presenting in the first two decades) include dermoid cysts, thyroglossal duct cysts, bronchogenic cysts, cystic hygromas, and branchial cleft cysts.[1,3,4,6]
Odontogenic cysts may have any clinical appearance, and they are usually found incidentally as multiple radiolucent lesions of the mandible. For the dermatologist, the findings of multiple odontogenic cysts are diagnostic for basal cell nevus syndrome until proven otherwise. It is estimated that approximately five percent of people with odontogenic cysts have basal cell nevus syndrome. Early identification of these patients and their lesions is key to improving long-term survival and quality of life.[3,4]
The majority of lesions seen by dermatologists are benign. However, some cyst-like masses may represent a more serious disorder, which must be evaluated carefully prior to surgical procedures.[2,6] This article presents seven mimickers of cysts in adults and reviews the clinical, histological, and surgical management of these lesions.
Nodular Hidradenomas
Nodular hidradenomas (NH) are benign adnexal tumors of eccrine or apocrine origin that may present as solitary nodules simulating epidermoid cysts. The most common site of involvement for NH is the head and neck (30% of patients), but they may occur in any site.[7] These neoplasms occur primarily in adults (women more than men) though children may be affected. NH have been described in the literature under a myriad of names, including clear cell myoepitheliomas, clear cell hidradenomas, eccrine acrospiromas, and eccrine spiradenomas.[7–9]
The diagnosis of these tumors on a clinical basis is extremely difficult. Clinically, these tumors present as painless, solitary, slowly enlarging, freely movable, dermal nodules measuring 1 to 6cm in diameter.[9] Some NH show superficial ulceration with serous discharge; others may have pedunculated or cystic appearance.
The diagnosis of NH is usually done by skin biopsy (Figure 1). Histologically, these lesions can simulate eccrine poromas and glomus tumors.[3,8] The differential diagnosis for these tumors may include hemangioma, lipoma, lymphangioma, squamous cell carcinoma (SCC), or basal cell carcinoma( BCC).[1,2,10]
Most cases of NH have a benign course. The recurrence rate of surgically excised tumors is approximately 10 percent, according to a large series published by Johnson et al.[7,8] Recurrence may occur in inadequately excised tumors and are usually located in the deeper dermis or subcutaneous fat.
Given the possibility of recurrence and the aggressive nature of their malignant counterpart, complete surgical excision of NH is recommended.[11] Malignant hidradenomas (hydradenocarcinomas) are rare and often originate de novo. These tumors are locally invasive and may also metastasize to the bone and lymph nodes.[7,9] The discrimination of NH from hidradenocarcinoma should be made histologically. The main histological features for malignancy are increased mitotic activity, the presence of angiolymphatic invasion, necrosis, local extension into deep tissues, and a dispersed growth pattern.[3,7,9] Furthermore, most NH are epithelial membrane antigen (EMA), CAM5.2, and vimentin positive as well as S-100 protein, and smooth muscle actin (SMA) negative.[1,3,7,8,10]
For benign NH, complete surgical excision is often curative. Mohs micrographic surgery (MMS) has been successfully performed in isolated cases of NH and is a good alternative approach in the treatment of large or recurrent tumors where wider subclinical spread is possible.[7,11]
Cutaneous B-cell Lymphoma
Cutaneous B-cell lymphomas compose a group of malignant B-cell lymphomas primarily affecting the skin. Although these neoplasms share morphological characteristics with nodal lymphomas, cutaneous B-cell lymphomas have different prognosis, natural history, and treatment protocols.[4,5]
Although there are different classification schemes reported in the literature, there are no general agreements as to a classification system.[3,4] Primary cutaneous lymphomas are defined by the European Organization for Research and Treatment of Cancer (EORTC) as, “lymphomas confined to the skin at presentation and without evidence of extra cutaneous spread for at least six months thereafter.”[12] This classification divides primary cutaneous B-cell lymphomas into three subtypes: indolent (follicle center lymphoma and immunocytoma), intermediate (large B-cell lymphoma of the leg) and provisional (intravascular large B-cell lymphoma and plasmacytoma).[12,13]
The clinical manifestations of cutaneous B-cell lymphomas vary: cutaneous follicle center cell lymphomas, the most common subtype (90% of all forms of cutaneous B-cell lymphomas), often occur as pink-to-plum-colored nodules primarily over the scalp, forehead, or back.[3,14] Immunocytomas, also known as cutaneous marginal zone B-cell lymphomas,[3] may present as red-to-brown nodules over the arms or trunk, while large B-cell lymphomas present as solitary or multiple papules, nodules, or tumors often over the lower extremities. Intravascular B-cell lymphomas should be considered part of the differential diagnosis of patients with dementia (or other neurological deficit), presenting with erythematous-to-blue nodules or plaques of the face or extremities. Lastly, primary cutaneous plasmacytomas most commonly affect elderly men and present as red-brown nodules over the head and trunk.[3,14,15]
Naturally, biopsy and systemic evaluation is imperative to establish the diagnosis (Figure 2).[3,13–15] Treatment is based on the type of B-cell lymphoma. In the authors’ case, excision, rituximab treatment, and local radiation after a negative systemic evaluation was effective.
Epithelioid Sarcoma
Epithelioid sarcomas (ES) are very rare, malignant, soft tissue neoplasms that present as slow-growing nodules favoring the distal extremities of young adults.[4,5,16] Many regard ES as the most common soft tissue sarcoma of the hand and wrist, followed by synovial cell sarcoma and malignant fibrous histiocytoma.[16] The pathogenesis of ES remains unknown. Recently, an N-ras oncogene mutation has been postulated as a possible initiating event in the formation of ES.[17] Some authors favor synovial origin while others postulate histiocytic, fibroblastic, or myofibroblastic cells as the origin of ES.[1,3,16]
Clinically, these tumors present as slow-growing, tan-white nodules with indistinct margins, typically involving the digits, the palm, or the volar forearm. The clinical presentation of ES may resemble giant cell tumor of the tendon sheath, nodular fasciitis, rheumatoid nodule, or a ganglion cyst.[4,17] Conversely, ES may present with focal areas of necrosis mimicking other malignant neoplasms, such as SCC, angiosarcoma, or malignant melanoma.[3,4,16] Given their macroscopic appearance, ES may be misdiagnosed and treatment is often delayed. Furthermore, the underlying ulceration and drainage may be confused with infection (cellulitis or abscess) or a ruptured cyst.
Histologically, ES present with large epithelioid (polygonal) cells with eosinophilic cytoplasm, round nuclei, and prominent nucleoli and focal areas of necrosis (Figure 3).[1,3,4,18] These tumors are differentiated from other soft tissue malignancies by immunohistochemistry. These tumors are positive for cytokeratin, EMA, and vimentin and negative for CD45 or S-100 protein.[1,3,4,16]
ES tend to be multifocal and infiltrative, with multiple reports of neural and fascial involvement.[16–19] Spread to the regional lymph nodes is a common finding. Tumor recurrence is seen frequently. Local recurrence develops in 80 percent (usually within six months of initial treatment) and metastasis in 30 to 45 percent (regional lymph nodes and lungs are the most common sites).[16,18]
Adverse prognostic factors corresponding to high recurrence rates include tumor size (>5cm), vascular invasion, numerous mitotic figures, necrosis of more than 30 percent, and axial location.[16] The five-year survival rate is 50 percent and metastases are common.[4,5,16]
Preoperative evaluation for suspicious lesions should include imaging studies with magnetic resonance imaging (MRI).[19] Treatment must include a wide radical excision with sentinel lymph node sampling. One study of 55 patients reviewed retrospectively has shown decreasing recurrence rate with increasing aggressiveness of initial surgical resection. External beam radiation, lymphadenectomy, or brachytherapy may be considered for local control of recurrent large lesions.
Merkel Cell Carcinoma
First described by Toker in 1972, Merkel cell carcinoma (MCC) is an aggressive neuroendocrine tumor with a propensity to develop early metastasis. Its cell of origin is thought to be the Merkel cell, derived from the neural crest and located in the basal layer of the epidermis.[4,5,20] Ultraviolet radiation, immunosuppression, and exposure to arsenic have all been postulated as possible etiologies.[4]
Given its susceptibility for early lymphatic invasion and nodal and hematogenous spread, MCC has been described as one of the most aggressive cutaneous malignancies.[20]
Clinically, MCC typically appears as a red or violaceous, firm, well-circumscribed nodule or plaque with overlying telangiectasias.[4,5] It usually arises from sun-exposed areas, such as the head and neck, but other sites have been reported, including the buttocks, distal extremities, and trunk.[20,21] Given its nonspecific clinical morphology, MCC may be mistaken for BCCs, amelanotic melanomas, SCCs or cutaneous lymphomas, and vascular proliferations, such as angiomas and angiosarcomas.[20–22]
Approximately one third of cases have involvement of the regional lymph nodes at the time of diagnosis (nodal status is a marker of distant spread), and hematogenous spread is cited to occur in about 50 percent of cases.[21] Other sites of metastasis include skin, lymph nodes, liver, bone, and brain.[20,21]
Histologically, MCC is challenging to diagnose, as it can sometimes mimic other poorly differentiated small cell tumors, such as small cell carcinoma of the lung, neuroblastoma, or lymphoma (Figure 4).[1,3] Immuno-histochemistry aids in distinguishing MCC from these other malignancies. MCC typically expresses both cytokeratin markers (CK20, CAM 5.2) as well as neuroendocrine markers, such as synaptophysin and neuron specific enolase and chromogranin A.[3,20–22] S-100 protein and thyroid transcription factor (TTF-1) are usually negative. These findings help in the differential diagnosis of melanoma and small cell carcinoma, respectively.[3–5,20–22]
There are no specific staging systems for MCC. The most commonly adopted system separates patients into three groups: Stage 1 (patients with localized disease), Stage 2 (regional disease), and Stage 3 (distant disease). Approximately 70 to 80 percent of patients will present with Stage 1 disease, 15 to 30 percent with Stage 2, and less than four percent with Stage 3 disease.[4,20,22]
The principal objective in the treatment of MCC is tumor control at the primary site and disease prevention. Treatment for Stages 1 and 2 involves surgery and radiation therapy (Stage 1, 5-year survival rates are cited at about 60–70%).[4,20] Given the tumor’s tendency to spread to local lymph nodes, wide surgical excisions with 2.5 to 3.0cm margins have been advocated.[4,20] MMS has been reported as an alternative option for surgical clearance. In 2002, Boyer et al reviewed 45 patients with MCC treated with MMS. Twenty five received MMS alone and 20 underwent MMS plus adjuvant radiotherapy. Of the 25 patients in the surgery group, four had evidence of recurrence; whereas, no recurrences were noted in the group that received adjuvant radiotherapy.[23] The role of prophylactic lymph node dissection is still controversial. Some surgeons recommend this procedure in patients with tumors with poor prognostic indicators (size larger than 2cm, small cell histological pattern, and mitotic rates of greater than 10 mitosis per high power field).[4,20,21] For patients with Stage 3 MCC, palliative care is commonly administered. Chemotherapy and radiation therapy are also administered; however, the median survival rate for Stage 3 patients is about 9 to 10 months.[20,22]
Cutaneous Metastasis
Cutaneous metastases are uncommon and may be the first clinical sign of an internal malignancy.[4] Dissemination is usually via hematogenous or lymphatic spread. Reports indicate that the incidence of metastasis to the skin occurs approximately in 1 to 2 percent of patients with internal malignancies.[1,4] Lookingbill et al performed a retrospective study of 4,020 patients with metastatic disease and found that 10 percent (402 patients) had cutaneous involvement.[24] Furthermore, in a second study, these authors looked at 7,316 patients with internal malignancies and found that two percent (367 patients) had evidence of metastatic skin involvement. In most cases, the development of cutaneous metastases represents a poor prognosis.[25] In fact, some reports refer to an average survival time of 3 to 6 months after the appearance of cutaneous metastases.[4,26]
Adenocarcinomas (from primary carcinomas of the large intestine, lung, or breast) represent approximately 60 to 70 percent of cutaneous metastases.[4,26] Squamous cell neoplasms (oral cavity, lung, and esophagus) represent approximately 15 percent of metastatic disease in the skin, with the remainder percentage involving malignant melanoma and other anaplastic tumors.[26,27]
Clinically, cutaneous metastases often present as multiple, nontender, dome-shaped nodules.[3,5] These lesions may be red, purple, or skin colored and can vary in size from 1 to 3cm. Often, these lesions tend to appear near the anatomical site of the primary tumor, but they can be found anywhere on the body, even at sites of diagnostic or surgical procedures.[4,5,26] A small percentage involve the scalp, and of those, lung and kidney in men and breast in women are the most prevalent.[26, 27]
Histologically, metastatic carcinoma of the skin is usually located in the mid-dermis with cells representing morphologies of the primary tumor.[1,3] The epidermis is usually normal except for cases of epidermotropic metastases as is the case of malignant melanoma.[3,27] Immunohistochemistry is helpful in the host diagnosis of cutaneous metastases: monoclonal antibodies against thyroglobulin, calcitonin, CD45, EMA, S-100 protein, cytokeratin, vimentin, and others are useful to confirm the etiology of the primary tumor.[1,3,4]
Granular Cell Tumor
This neoplasm usually presents as a solitary, asymptomatic, well-circumscribed, skin-colored nodule. Most commonly, granular cell tumors are found in the head and neck with approximately 30 percent present on the oral cavity, especially the tongue.[4,5,28] Recent evidence suggests that granular cell tumors are neurally derived (Schwann cells), although lately this has been a source of controversy.[4,5,29]
Histologically, the cells are pale and polygonal, with a very distinct, coarsely granular cytoplasm (Figure 5).[3] The granules seen on microscopy are secondary to the accumulation of lysosomal granules on the cytoplasm.[1,3] Malignant granular cell tumors are very rare with only a handful of cases reported involving the skin. Excisional surgery is the treatment of choice.[4,5,28]
Cutaneous Meningiomas
Cutaneous meningiomas are very rare tumors and represent a group of neoplasm arising from the meninges.[3] The histogenesis of cutaneous meningiomas is the meningo-epithelial cell, which is ectopically present in the dermis or subcutaneous tissue.[30] The majority of these tumors arise on the occipital scalp or along the cranial suture lines. These tumors should be considered in the differential diagnosis of a firm, subcutaneous lesion arising on the scalp or prevertebral region.[3,31]
Lopez et al classified cutaneous meningiomas into three groups[32]: Type 1 (primary cutaneous meningiomas) is almost always congenital, and tumors arise from ectopic arachnoid cells located outside the calvarium as a result of abnormal bone development. Clinically, they may resemble epidermoid cysts, nevi, or areas of alopecia areata and diagnosis on histological markers are usually incidental.[30,32] Type 2 (meningioma of soft tissue with skin extension) occurs most commonly on the forehead, scalp, mandible, and neck (along distribution of cranial and spinal nerves). These types of tumors are more common in adults and grow slowly over a number of months to years.[30,32] Type 3 (central nervous system meningioma with extension into the skin) can spread to the skin through bony defects (after traumatic injury) or by hematogenous or lymphatic dissemination. They are aggressive and treatment involves tumor staging and extensive surgery.[30–33]
Histologically, these tumors are well circumscribed, with nests of spindle-shaped cells intermixed with fibrocollagenous stroma. Psammoma bodies may be present.[1,3,30]
Types 1 and 2 lesions are usually excised as they can grow and be disfiguring or compress adjacent structures.[30,31] In the preoperative evaluation, a CT scan of the head to rule out intracranial connection is essential. Once proven, if there is no intracranial or intravertebral connection, the lesion can be treated conservatively unless the size is significant or pressure symptoms supervene.[30,31] Intracranial or intravertebral connection indicates spread from a primary lesion. In this case, prognosis is poor.
Conclusion
Cysts and cyst mimickers are common occurrences in dermatology. Although many can be treated by simple excisional surgery, a careful evaluation on any cyst-like structure may reveal different clinical, etiological, embryological, or histological findings.
In many instances, these lesions may represent a more complex clinical picture requiring further workup by other sub-specialists before adequate surgical treatment. A thorough understanding of other lesions that appear to be cysts is important to the dermatologist and can guide his or her approach to this group of cutaneous masses.
References
1. Elder D, Elenitsas R, Jaworsky C, Johnson B. Lever’s Histopathology of the Skin, 8th ed. Philadelphia: Lippincott Williams & Wilkins; 1997
2. Baldwin HE, Berck CM, Lynnfield YL. Subcutaneous nodules of the scalp: preoperative management. J Am Acad Dermatol. 1991;25(5 Pt 1):819–830.
3. Weedom, D. Skin Pathology. 2nd ed. London: Churchill Livingstone; 2002.
4. Bolognia J, Jorizzo J, Rapini R. Dermatology. New York: Mosby; 2004.
5. Odom R, James W, Berger T. Andrews’ Diseases of the Skin. Clinical Dermatology. 9th ed. Philadelphia: Saunders; 2000.
6. Jaworsky C, Nguyen P, Werth VP. Mimickers of cysts. J Am Acad Dermatol. 1993;29(2 Pt 1):260–262.
7. Hernandez-Perez E, Cestoni-Parducci R .Nodular hidradenoma and hidradenocarcinoma. A 10-year review. J Am Acad Dermatol. 1985;12:15–20.
8. Haupt HM, Stern JB, Berlin SJ. Immunohistochemistry in the differential diagnosis of nodular hidradenoma and glomus tumor. Am J Dermatopathol. 1992;14(4):310–314.
9. Ohta M, Hiramoto M, Fujii M, Togo T. Nodular hidradenocarcinoma on the scalp of a young woman: case report and review of literature. Dermatol Surg. 2004;30(9):1265–1268.
10. Domoto H, Terahata S, Sato K, Tamai S. Nodular hidradenoma of the breast: report of two cases with literature review. Pathol Int. 1998;48(11):907–911.
11. Vodovnik A. Co-expression of S-100 and smooth muscle actin in nodular hidradenoma. Am J Dermatopathol. 2003;25(4):361–362.
12. Slater DN. The new World Health Organization-European Organization for Research and Treatment of Cancer classification for cutaneous lymphomas: a practical marriage of two giants. Br J Dermatol. 2005;153(5):874–880.
13. Burg G, Kempf W, Cozzio A, et al. WHO/EORTC classification of cutaneous lymphomas 2005: histological and molecular aspects. J Cutan Pathol. 2005;32(10):647–674.
14. Storz MN, van de Rijn M, Kim YH, et al. Gene expression profiles of cutaneous B-cell lymphoma. J Invest Dermatol. 2003;120(5):865–870.
15. Kerl H, Kodama K, Cerroni L. Diagnostic principles and new developments in primary cutaneous B-cell lymphomas. J Dermatol Sci. 2004;34(3):167–175.
16. Murray PM. Soft tissue sarcoma of the upper extremity. Hand Clin. 2004;20(3):325–333.
17. Kagami S, Saeki H, Idezuki T, et al. Epithelioid sarcoma associated with lung adenocarcinoma. J Dermatol. 2005;32(11):904–908.
18. Zimmer LA, Gillman G, Barnes L. Postauricular epithelioid sarcoma. Otolaryngol Head Neck Surg. 2004;131(6): 1022–1023.
19. Chao KC, Chen C, Hsieh SC, et al. MRI of epithelioid sarcoma of the thigh. Clin Imaging. 2005;29(1):60–63.
20. Poulsen M. Merkel cell carcinoma of skin: diagnosis and management strategies. Drugs Aging. 2005;22(3):219–229.
21. Acebo E, Vidaurrazaga N, Varas C, Burgos-Bretones JJ, Diaz-Perez JL. Merkel cell carcinoma: a clinicopathological study of 11 cases. J Eur Acad Dermatol Venereol. 2005;19(5):546–551.
22. Boyse K, Foley EH, Bradley V, Scarborough D. Merkel cell carcinoma: a case report with treatment summary and updates. Cutis. 2004;74(6):350–356.
23. Boyer JD, Zitelli JA, Brodland DG, D’Angelo G. Local control of primary Merkel cell carcinoma: review of 45 cases treated with Mohs micrographic surgery with and without adjuvant radiation. J Am Acad Dermatol. 2002;47(6):885–992.
24. Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4,020 patients. J Am Acad Dermatol. 1993;29(2 Pt 1):228–236.
25. Lookingbill DP, Spangler N, Sexton FM. Skin involvement as the presenting sign of internal carcinoma. A retrospective study of 7,316 cancer patients. J Am Acad Dermatol. 1990;22(1):19–26.
26. Brownstein MH, Helwig EB. Patterns of cutaneous metastasis. Arch Dermatol. 1972;105(6):862–868.
27. Brownstein MH, Helwig EB. Metastatic tumors of the skin. Cancer. 1972;29(5):1298–1307.
28. Moon CM, Smith SB, Biediger TL. What is your diagnosis? Granular cell tumor. Cutis. 2005;75(1):21,23–24.
29. Arican O, Ciralik H, Sasmaz S. Multiple plaques on the back: S-100 negative benign granular cell tumor. J Dermatol. 2005;32(7):585–588.
30. Gelli MC, Pasquinelli G, Martinelli G, Gardini G. Cutaneous meningioma: histochemical, immunohistochemical and ultrastructural investigation. Histopathology. 1993;23(6): 576–578.
31. Ragoowansi R, Thomas V, Powell BW. Cutaneous meningioma of the scalp: a case report and review of literature. Br J Plast Surg. 1998;51(5):402–404.
32. Lopez DA, Silvers DN, Helwig EB. Cutaneous meningiomas- a clinicopathologic study. Cancer. 1974;34(3):728–744.
33. Kakizoe S, Kojiro M, Hikita N. Primary cutaneous meningioma. Report of a case. Acta Pathol Jpn. 1987;37(3):511–514.