Laura Englander, MD; Jason J. Emer, MD; Danielle McClain, MD;
Bijal Amin, MD; Ryan B. Turner, MD
Laura Englander, MD and Ryan B. Turner, MD are from Department of Dermatology, Albert Einstein School of Medicine, Bronx, New York; Jason J. Emer, MD is from Department of Dermatology,
Mount Sinai School of Medicine, New York, New York; Danielle McClain, MD is from Department of Pathology, Albert Einstein School of Medicine, Bronx, New York
Authors’ note: Drs. Englander and Emer contributed equally to this work
Disclosure: The authors report no relevant conflicts of interest.
Abstract
Eccrine spiradenoma is a benign adnexal neoplasm that has been historically designated as a tumor of eccrine differentiation, although current reconsideration indicates an apocrine process. It usually presents on the trunk and extremities as a tender dermal or subcutaneous papule or nodule frequently with a pink or blue hue. The clinical picture is often not distinct and biopsy is required for diagnosis. Eccrine spiradenoma can present in a variety of ways, including as tumors arranged in zosteriform/dermatomal and/or blaschkoid distributions, often precluding a straightforward diagnosis. Proper diagnosis of eccrine spiradenoma is important due to the occurrence of potentially life-threatening malignant transformation. This article illustrates a rare presentation of eccrine spiradenoma with a concise review for the dermatologist. (J Clin Aesthet Dermatol. 2011;4(4):38–44.)
Eccrine spiradenoma (ES), first described in 1956, is an uncommon, benign, dermal tumor of apocrine differentiation derived from cutaneous sweat glands.[1] It classically presents in the 2nd to 4th decades of life as a small, painful, grey to pink nodule on the upper ventral aspect of the body. Most presentations of ES are solitary, comprising more than 97 percent of cases. The incidence of ES is roughly equivalent in men and women.[2–4] Multiple ES is a rare phenomenon, comprising less than two percent of all cases and appears to be predominant among women.[5,6] Approximately 37 cases of multiple ES, including those in zosteriform/dermatomal and/or blaschkoid distributions, have been reported in the literature.[2,4,6–28] Given the rarity of multiple ES in a linear distribution and the uncommon risk of malignant transformation, the authors present this case for interest, review, and awareness.
Case Report
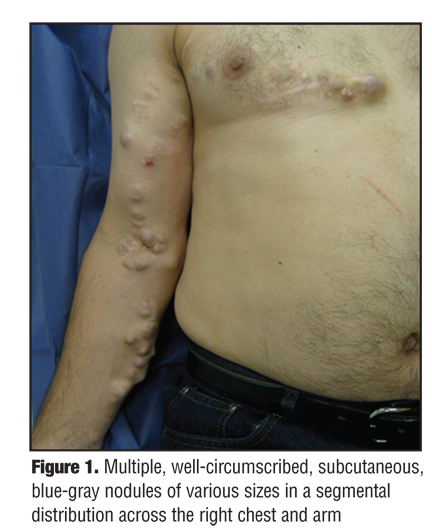
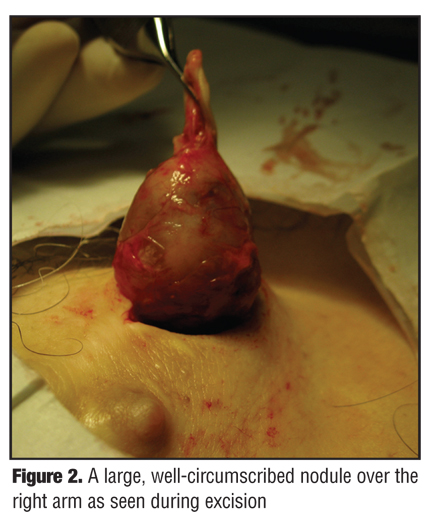
A 55-year-old Caucasian man presented to the authors’ outpatient dermatology clinic complaining of a 20-year history of painful lesions on the right chest and arm. Of patient concern, the lesions were recently more painful and had increased in size and number. His past medical history was unremarkable. He denied any family members with similar skin findings or any history of skin cancers. Physical exam revealed multiple, well-circumscribed, subcutaneous, blue-grey nodules in a dermatomal distribution across the right chest and arm (Figure 1). The nodules were tender, firm, and fixed to the overlying skin and ranged in size from 0.5 to 6cm in diameter. Surgical excision of the most prominent, painful nodule of the right arm was sent for histological analysis (Figure 2).
{kind=link}
{kind=link}
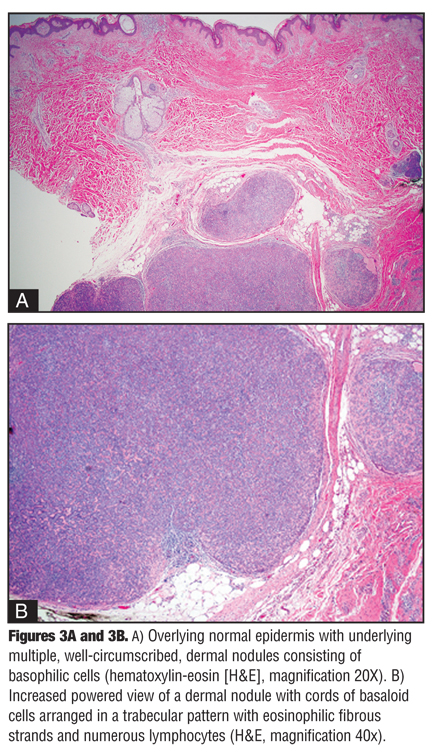
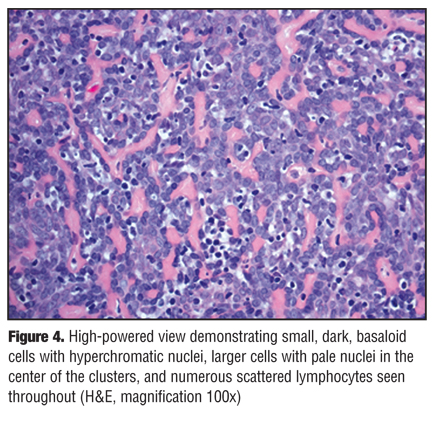
Histological findings demonstrated sharply delineated dermal nodules comprising basaloid cells arranged in a trabecular pattern surrounded by eosinophilic fibrous strands (Figure 3). Small, dark, basaloid cells with hyperchromatic nuclei, larger cells with pale nuclei in the center of the clusters, and numerous scattered lymphocytes were also seen throughout, consistent with a diagnosis of ES (Figure 4). Typical spiradenoma features, as well as small areas with a cylindroma-like “jigsaw puzzle” pattern, were also appreciated. No evidence of malignant transformation was noted.
The patient underwent surgical excision with complex, linear closure of multiple chest and arm nodules given the significant pain experienced while wearing a seatbelt during driving. Excellent cosmetic and functional results were seen and the patient was advised to return for future excisions in the case of increasingly painful or rapidly growing lesions. While there was no evidence of malignant transformation in this patient, continuous follow up is necessary given the rapid evolution and possibility of malignant transformation within long-standing lesions.
{kind=link}
{kind=link}
Discussion
Introduction. Few cases of multiple ES have been reported in the literature to date. The present case is a unique presentation of multiple ES in a pattern following the T1 dermatome (Figure 1). In cases of multiple ES, it has been proposed that an abnormal clone of multipotent stem cells of the folliculosebaceous-aprocrine unit arise during embryogenesis subsequently producing a proliferation of abnormal cells resulting in nodule formation and possible malignant transformation.[29–33] Histological findings are often consistent with ES and cylindroma within the same biopsy as many tumors show overlapping features of these two entities.[12] It may be that these two entities represent two extremes on a continuous spectrum of dermal tumor originating from a common progenitor.[34] Spiradenomas occurring in multiplicity and in concert with cylindroma or trichoepitheliomas should prompt investigation into a diagnosis of Brooke-Spiegler syndrome.
Brooke-Spiegler syndrome is an uncommon autosomal dominant disorder characterized by a high affinity to form multiple adnexal neoplasms, especially trichoepitheliomas, cylindromas, and spiradenomas.[35] The occurrence of both follicular and apocrine differentiation may represent separate paths of maturation from a single germ cell, explaining how several types of tumor coexist. In fact, germline mutations in the cylindromatosis (CYLD) gene have been described in families with cylindromas, trichoepitheliomas, and spiradenomas.[36] Recently, the CYLD gene has been recognized as an important tumor suppressor gene with a prominent role in the regulation of nuclear factor kappa-beta (NF-kb), a transcription factor that promotes cell survival and oncogenesis.[37,38] Thus, defective tumor suppression genes seem to be important in the development of spiradenomas.
Benign ES generally comprises lesions ranging from 0.5 to 3cm in diameter that remain chronically stable.[39,40] Malignant transformation is rare and occurs more often in cases of multiple benign ES than in solitary cases.[39]
Malignant ES, first described in 1972, is thought to arise from transformation of a long-standing, benign ES.[30] De-novo cases have recently been reported in the literature.[41] No age, gender, or site predilection has been described, and malignant transformation generally occurs 20 to 30 years after initial lesion detection.[39] Many mechanisms have been proposed, such as trauma, but none are substantiated in the literature.[42–44] In these rare cases of malignant potential, p53 may be increased even in the presence of a benign-appearing histology.[45,46] Malignant ES are aggressive tumors that spread both hematogenously and lymphogenously and have been reported to have a mortality of 39 percent if left untreated.[43,44] Since the rate of metastasis is close to 50 percent and can result in death, these patients must be followed closely. Currently, no guidelines have been developed for surveillance in patients with multiple ES, although it seems reasonable that patients with larger-sized, multiple, and/or symptomatic lesions be monitored regularly.
Etiology. The exact etiology of ES still remains controversial. ES has never been observed on glabrous skin, as it is an undifferentiated or poorly differentiated benign adnexal neoplasm historically designated as a tumor of eccrine lineage. However, both ES and cylindroma can sometimes display tubular differentiation with a “decapitation” pattern as would be expected in an apocrine neoplasm.[47,48] Thus, recent assessment has indicated an apocrine process. As previously described, ES commonly occurs jointly with other cutaneous appendage tumors, such as cylindroma, trichoepithelioma, and trichoblastoma, which points toward the development of folliculosebaceous-apocrine lineage rather than eccrine differentiation.[49,50] Further, if ES were truly eccrine, occurrence on the palm or sole would be paramount rather than rarely seen. In Brooke-Spiegler syndrome, a defective tumor suppressor gene, CYLD on chromosome 16q, results in numerous cutaneous appendage tumors suggesting kinship rather than disparate lineages.[46,51] The most recent hypothesis is suggestive of an abnormal multipotent stem cell in the folliculosebaceous-apocrine unit, although some reports have suggested trauma as the inciting factor.[43,52] p53 expression in malignant ES is also suggestive of an underlying abnormality of tumor suppression or innate surveillance, but the significance of this observation is currently unclear.
Presentation. ES classically presents as a dermal or subcutaneous papule or nodule that can range in size up to many centimeters. A pink or blue hue overlying the nodule is suggestive, but clinical diagnosis alone cannot give accurate diagnosis. ES are often painful and are in the differential diagnosis of painful dermal tumors (as described by Naversen et al[53]) characterized by the popular pneumonic “LEND AN EGG,” which stands for leiomyoma, ES, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomus tumor, and granular cell tumor.[54]
No clear categorization exists for the multitude of presentations of ES. Rarely, ES presents as multifocal or multiple localized tumors arranged in a linear, zosteriform, or nevoid distribution. A recent case report and review proposed a system of categorization based on both clinical and histological findings.[6] Clinically, they defined the lesions as solitary or multiple, with further distinction to describe the clinical appearance and define the distribution/pattern (linear, zosteriform, nevoid, or blaschkoid). Histologically, the authors focused on determining whether the lesions were benign or malignant, and they identified three histological classifications: common, vascular, and cystic.
A simple system of clinical characterization comprises descriptors based on quantity, location, and distribution. Quantity indicates whether the lesion is solitary or in multiplicity. Location denotes whether the presentation is multifocal, present at various body sites, or localized and isolated to a particular area. Distribution describes how the lesions are configured or arranged in the previously described location. Distribution can be zosteriform/ dermatomal, blaschkoid, or linear. Zosteriform is a term used to describe lesions that resemble herpes zoster, which often follows a dermatomal distribution. Dermatomal refers to a distribution along a dermatome (or series of dermatomes) corresponding to an area of skin innervated by site-specific spinal nerves. Thus, zosteriform and dermatomal are similar and together refer to a band-like unilateral skin lesion along a cutaneous distribution of a spinal nerve(s). In contrast, a blaschkoid pattern follows the lines of Blaschko, a surface pattern invisible under normal conditions and thought to represent pathways of epidermal cell migration and proliferation during embryological development (genetic mosaicism). They follow a “V” shape over the back and an “S” shape over the chest, stomach, abdomen, and head/neck and appear as whorls typically seen in pigmentary disorders, X-linked genetic disorders, or acquired inflammatory skin conditions. In the literature, the terms “blaschkoid” and “dermatomal” have been mistakenly used interchangeably. Lines of Blaschko do not correspond to nervous, muscular, or lymphatic systems, and dermatomal lines are never in a whorled pattern. Linear is used to describe any lesions in a straight line system and is the most generic of the descriptors.
Diagnosis. The definitive diagnosis of ES consists of a skin biopsy; however, fine needle cytology has also been reported.[55] ES comprises large, sharply circumscribed, basophilic nodules (“cannon balls” or “blue balls”) arranged in intertwining cords, islands, or sheets in the dermis or subcutaneous tissue that are surrounded by a fibrous capsule.[56] Basaloid cells can be composed of two distinct morphologies with one cell type that is larger, pale, and with an ovoid nuclei and the other type that is smaller, dark, and with a compact hyperchromatic nuclei. Duct-like structures can be present at the center of the lesions, which often have scattered lymphocytes.[4] Strands of cells are positive for cytokeratin and the lumina are positive for carcinoembryonic antigen.[57] S-100 staining has been noted in ES and cylindromas attributed to eccrine differentiation. In cases where ES has co-expression of cytokeratin and smooth muscle actin, authors have suggested differentiation toward myoepithelial cells.[58]
Malignant ES is suspected in patients with rapid enlargement, an increase in number, change in color, or with the development of symptoms, such as pain, ulceration, or pruritus. Histological findings may include atypical cells, elevated mitotic counts, loss of the typical lobular pattern and dual cell population, rare foci of necrosis, and enlarged cells of one cell type on histological evaluation.[2,44] Additionally, tumor cells extending through the capsule into adjacent stroma are also suggestive.[59]
Immunohistochemistry can be useful in evaluating for malignant cases of ES. In a case of malignant ES of the breast, typical histological features were noted along with areas of adenocarcinoma, squamous cell carcinoma, and sarcoma with staining positive for the p53 protein.[60] Others have demonstrated that cells may express cytokeratins, epithelial membrane antigen, and p53.[41]
In 2000, Granter et al[61,62] divided malignant ES into two groups (high and low grade) based on clinical and histological features. High-grade malignant ES has more obvious features of malignancy with pleomorphism and increased mitoses (4–32/hpf).[44,61] Low-grade malignant ES has less obvious features of malignancy, such as loss of the typical ductal cells, mild-to-moderate pleomorphism, and lower mitotic rates (2–10/hpf).[44,61] As explained previously, malignant ES tends to have a high rate of metastasis to regional lymph nodes, lungs, brain, and liver; therefore, radiological studies, such as x-ray, computed tomography (CT), or magnetic resonance imaging (MRI), are useful in assessing for metastatic foci.
Differential diagnosis. The diagnosis of ES can be elusive given its multiple presentations and frequent lack of skin surface changes. Correct diagnosis is critical due to the potential for malignancy. Popular pneumonics are used to describe painful dermal tumors as described above and these should all be taken into consideration on initial evaluation. While the following additional entities are not generally painful, trichoepithelioma, lipoma, cylindroma, and poroma should be considered in the differential given their similar presentations. Further, in long-standing lesions and those with symptoms or recent growth, malignant eccrine poroma, malignant nodular hidradenoma, and chronic ulcers should be considered depending on the presentation. One case report documents a toe ulcer followed for four years as a chronic nonhealing ulcer, but later found to be a malignant ES.[63]
The diagnosis can generally be easily distinguished histologically if the clinical picture is not distinctive. However, histological differentiation of cylindroma and ES is less straightforward. As a general rule, cylindromas tend to present on the head and neck and are usually not painful as compared to ES, which present on the trunk and extremities and are generally painful. Histologically, cylindroma is differentiated by the presence of basaloid cells arranged in “islands” that fit together like a jigsaw puzzle, rather than in “rosettes” as seen in ES.[64,65] Other histological distinctions include the presence of a densely eosinophilic PAS-positive material present around the tumor “islands” in the case of cylindroma, as well as the absence of scattered lymphocytes that are often present in ES.
Treatment and management. The mainstay of treatment for both benign and malignant ES is surgical removal, although this may not be necessary unless the lesions are cosmetically intolerable, painful, or increasing in size or number. Other options, such as radiotherapy, carbon dioxide laser ablation, or chemotherapy, are reserved for cases in multiplicity or that are malignant. Due to the potential for malignant transformation, wide surgical excision or Mohs micrographic surgery offers the most conservative treatment choice, and lesions treated tend not to recur. One author suggests a combination of surgical management (staged surgical resections with reconstruction using complex linear closures and random-pattern cutaneous flaps) and medical management (carbon dioxide laser) for good cosmetic results.[34]
For the treatment of malignant ES, no treatment has been routinely established as paramount. Resection with wide margins (1–3cm) down to the fascia with lymph node dissection has been described as the most accepted treatment.[41,66,67] Radiation and chemotherapy have also been proposed, but no conclusive studies have substantiated optimal practice.[39,43,66] Hyperthermic limb perfusion chemotherapy has been tried as some believe that alterations of temperature may target treatment to tumor cells over healthy ones.[68] A recent meta-analysis of malignant ES demonstrated that patients with no metastasis who underwent surgical excision had a disease-free survival rate of 100 percent at 33 months.[66] In contrast, patients with distant metastases treated with either surgery alone or surgery and adjuvant chemotherapy had a median survival of 12 and 20 months, respectively. Despite the difference in median survival rates between the two groups, the results were not statistically significant. Resection of lymph node metastasis resulted in patients who were disease free at the final follow-up evaluation (mean 47 months) suggesting that, in the case of positive lymph nodes, surgical resection is certainly warranted. The authors concluded that aggressive surgical removal is necessary in cases of malignant ES with or without metastasis.[66]
In cases of malignant ES, close clinical follow up every three months the first year after resection, every six months the second year after resection, and annually thereafter is important given the possibility of recurrences. Due to the high rate of metastasis to the liver and lung, annual chest radiographs, liver function tests, liver ultrasound, chest and abdomen CT, and/or MRI may be useful.[44] But again, no consensus has validated these practices. Genetic counseling for cases of familial ES is also advised.[34]
Conclusion
ES is a dermal tumor of the sweat glands and its etiology is yet to be clearly identified. ES may present de novo or congenitally in clinically diverse presentations. The diagnosis is very important because of the potential for malignant transformation, especially in cases of multiple or symptomatic lesions. While extremely rare, malignant ES can be lethal if undiagnosed and untreated, and a high index of suspicion is needed on any benign lesion that rapidly changes in characteristics or is consistent with long-standing disease. Treatments for ES have not been well established, but surgical excision is currently the gold standard with low rates of recurrence. Case reports and reviews describing interesting presentations of ES are important to maintain awareness and to help guide proper management.
Acknowledgment
The authors would like to thank Brian Marciniak for his help in reviewing and editing this manuscript.
References
1. Kersting DW, Helwig EB. Eccrine spiradenoma. AMA Arch Derm. 1956;73(3):199–227.
2. Nath AK, Kumari R, Thappa DM. Eccrine spiradenoma with chondroid syringoma in Blaschkoid distribution. Indian J Dermatol Venereol Leprol. 2009;75(6):600–602.
3. Bedlow AJ, Cook MG, Kurwa A. Extensive naevoid eccrine spiradenoma. Br J Dermatol. 1999;140(1):154–157.
4. Ekmekci TR, Koslu A, Sakiz D. Congenital blaschkoid eccrine spiradenoma on the face. Eur J Dermatol. 2005;15(2): 73–74.
5. Gupta S, Jain VK, Singh U, Gupta S. Multiple eccrine spiradenoma in zosteriform distribution in a child. Ped Dermatol. 2000;17(5):384–386.
6. Yoshida A, Takahashi K, Maeda F, Akasaka T. Multiple vascular eccrine spiradenomas: a case report and published work review of multiple eccrine spiradenomas. J Dermatol. 2010;37(11):990–994.
7. Alfonso-Trujillo I, Arteaga-Hernández E, Pérez-Suárez JC. [Eccrine spiradenoma in a zosteriform distribution: presentation of a case]. Actas Dermosifiliogr. 2009;100(7): 619–620.
8. Rodríguez-Martín M, Sánchez González R, Sáez-Rodríguez M, et al. An unusual case of congenital linear eccrine spiradenoma. Pediatr Dermatol. 2009;26(2):180–183.
9. Lobo I, Amorim I, Selores M. Multiple nodules of the head—clinicopathologic challenge. Multiple eccrine spiradenoma. Int J Dermatol. 2009;48(3):237–238.
10. Agarwal S, Khanna R, Arya NC, Khanna AK. Malignant eccrine spiradenoma: an unusual presentation. Indian J Dermatol Venereol Leprol. 2002;68(5):290–291.
11. Han YD, Huan Y, Deng JL, et al. MRI appearance of multiple eccrine spiradenoma. Br J Radiol. 2007;80(949):e27–e29.
12. Bumgardner AC, Hsu S, Nunez-Gussman JK, Schwartz MR. Trichoepitheliomas and eccrine spiradenomas with spiradenoma/cylindroma overlap. Int J Dermatol. 2005;44(5): 415–417.
13. Yoshida A, Sato T, Sugawara Y, et al. Two cases of multiple eccrine spiradenoma with linear or localized formation. J Dermatol. 2004;31(7):564–548.
14. Altinyazar HC, Kargi E, Ozen O, et al. Multiple eccrine spiradenoma in zosteriform distribution. Plast Reconstr Surg. 2003;112(3):927–928.
15. Gupta S, Radotra BD, Kaur I, et al. Multiple linear eccrine spiradenomas with eyelid involvement. J Eur Acad Dermatol Venereol. 2001;15(2):163–166.
16. Kawakubo Y, Okano M, Iizuka M, Ohkido M. [Multiple eccrine spiradenoma. Histological association with dermal cylindroma]. Hautarzt. 1995;46(9):651–655.
17. Husek K, Plchová M, Samoh?l J. [Malignant transformation in multiple eccrine spiradenoma]. Cesk Patol. 1992;28(1): 48–52.
18. Bourrat E, Théodore-Lefebvre C, Beltzer-Garelly E, et al. [Multiple eccrine spiradenoma with Blaschko’s lines distribution]. Ann Dermatol Venereol. 1992;119(11): 897–898.
19. Wright S, Ryan J. Multiple familial eccrine spiradenoma with cylindroma. Acta Derm Venereol. 1990;70(1):79–82.
20. Revis P, Chyu J, Medenica M. Multiple eccrine spiradenoma: case report and review. J Cutan Pathol. 1988;15(4): 226–229.
21. Ikeya T. Multiple linear eccrine spiradenoma associated with multiple trichoepithelioma. J Dermatol. 1987;14(1):48–53.
22. Tsur H, Lipskier E, Fisher BK. Multiple linear spiradenomas. Plast Reconstr Surg. 1981;68(1):100–102.
23. Scheinfeld NS, Tarlow MM, Burgin S. Blaschkoid eccrine spiradenomas. Cutis. 2002;70(1):73–75.
24. Criton A, Aravindan KP. Zosteriform network of spiradenoma. Indian J Dermatol Venereol Leprol. 1996;62(3):185–186.
25. Ohtsuki Y, Ohtsuka H, Kurabayashi A, et al. Immuno-histochemical and electron microscopic studies of Langerhans cells in a case of multiple eccrine spiradenomas. Med Mol Morphol. 2007;40(4):221–225.
26. Noto G, Bongiorno MR, Pravata G, Arico M. Multiple nevoid spiradenomas. Am J Dermatopathol. 1994;16(3):280–284.
27. Shelley WB, Wood MG. A zosteriform network of spiradenoma. J Am Acad Dermatol. 1980;2(1):59–62.
28. Berberian BJ, Sulica VI, Kao GF. Familial multiple eccrine spiradenomas with cylindromatous features associated with epithelioma adenoides cysticum of Brooke. Cutis. 1990;46(1):46–50.
29. Clarke J, Ioffreda M, Helm KF. Multiple familial trichoepitheliomas: a folliculosebaceous-apocrine geno-dermatosis. Am J Dermatopathol. 2002;24(5):402–405.
30. Dabska M. Malignant transformation of eccrine spiradenoma. Pol Med J. 1972;11(2):388–396.
31. Ishikawa M, Nakanishi Y, Yamazaki N, Yamamoto A. Malignant eccrine spiradenoma: a case report and review of the literature. Dermatol Surg. 2001;27(1):67–70.
32. Chou SC, Lin SL, Tseng HH. Malignant eccrine spiradenoma: a case report with pulmonary metastasis. Pathol Int. 2004;54(3):208–212.
33. Engel CJ, Meads GE, Joseph NG, Stavraky W. Eccrine spiradenoma: a report of malignant transformation. Can J Surg. 1991;34(5):477–480.
34. Ter Pooten MC, Barrett K, Cook J. Familial eccrine spiradenoma: a case report and review of the literature. Dermatol Surg. 2003;29(4):411–414.
35. Uede K, Yamamoto Y, Furukawa F. Brooke-Spiegler syndrome associated with cylindroma, trichoepithelioma, spiradenoma, and syringoma. J Dermatol. 2004;31(1):32–38.
36. Blake PW, Toro JR. Update of cylindromatosis gene (CYLD) mutations in Brooke-Spiegler syndrome: novel insights into the role of deubiquitination in cell signaling. Hum Mutat. 2009;30(7):1025–1036.
37. Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17(1):1725–1734.
38. Kazakov DV, Schaller J, Vanecek T, et al. Brooke-Spiegler syndrome: report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors. J Cutan Pathol. 2010;37(8):886–890.
39. Braun-Falco M, Bonel H, Ring J, Hein R. Linear spiradenoma with focal malignant transformation. J Eur Acad Dermatol Venereol. 2003;17(3):308–312.
40. Cooper PH, Frierson HF Jr, Morrison AG. Malignant transformation of eccrine spiradenoma. Arch Dermatol. 1985;121(11):1445–1448.
41. Yildirim S, Akoz T, Akan M, Ege GA. De-novo malignant eccrine spiradenoma with an interesting and unusual location. Dermatol Surg. 2001;27(4):417–420.
42. Biernat W, Wosniak L. Spiradenocarcinoma: a clinico-pathologic and immunohistochemical study of three cases. Am J Dermatopathol. 1994;16(4):377–382.
43. Shaikh-Naidu N, Breitbart A. Eccrine spiradenoma of the upper extremity: case report and an algorithm for management. Eur J Plast Surg. 2003;26(3):160–163.
44. Hantash BM, Chan JL, Egbert BM, Gladstone HB. De-novo malignant eccrine spiradenoma: a case report and review of the literature. Dermatol Surg. 2006;32(9):1189–1198.
45. Dijkhuizen T, van den Berg E, Nikkels PG, et al. Cytogenetics of a case of eccrine spiradenoma. Hum Pathol. 1992;23(9): 1085–1087.
46. Kazakov DV, Grossmann P, Spagnolo DV, et al. Expression of p53 and TP53 mutational analysis in malignant neoplasms arising in pre-existing spiradenoma, cylindroma, and spiradenocylindroma, sporadic or associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2010;32(3): 215–321.
47. Hashimoto K, Lever WF. Histogenesis of skin appendage tumors. Arch Dermatol. 1969;100(3):356–369.
48. Lian F, Cockerell CJ. Cutaneous appendage tumors: familial cylindromatosis and associated tumors update. Adv Dermatol. 2005;21:217–234.
49. Goette DK, McConnell MA, Fowler VR. Cylindroma and eccrine spiradenoma coexistent in the same lesion. Arch Dermatol. 1982;118(4):273–274.
50. Michal M, Lamovec J, Mukensnabl P, Pizinger K. Spira-denocylindromas of the skin: tumors with morphological features of spiradenoma and cylindroma in the same lesion: report of 12 cases. Pathol Int. 1999;49(5):419–425.
51. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27(1): 27–33.
52. Arslan E, Unal S, Cinel L, Demirfan F, et al. Malignant eccrine spiradenoma occurring on a traumatized area. Plast Reconstr Surg. 2002;110(1):365–367.
53. Naverson DN, Trask DM, Watson FH, Burket JM. Painful tumors of the skin: “LEND AN EGG.” J Am Acad Dermatol. 1993;28(2 Pt 2):298–300.
54. Apatenko AK, Turusov VS. The painful tumours of the skin. Neoplasma. 1968;15(2):187–202.
55. Bosch MM, Boon ME. Fine-needle cytology of an eccrine spiradenoma of the breast: diagnosis made by a holistic approach. Diagn Cytopathol. 1992;8(4):366–368.
56. Park JW, Namkoong S, Chung J, et al. A case of eccrine spiradenoma in a patient with neurofibromatosis. Ann Dermatol. 2010;22(2):191–193.
57. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19(2):154–161.
58. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrine spiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21(1):121–123.
59. Tanese K, Sato T, Ishiko A. Malignant eccrine spiradenoma: case report and review of the literature, including 15 Japanese cases. Clin Exp Dermatol. 2009;35(1):51–55.
60. Saboorian MH, Kenny M, Ashfaq R, Albores-Saavedra J. Carcinosarcoma arising in eccrine spiradenoma of the breast. Report of a case and review of the literature. Arch Pathol Lab Med. 1996;120(5):501–504.
61. Granter SR, Seeger K, Calonje E, et al. Malignant eccrine spiradenoma (spiradenocarcinoma): a clinicopathologic study of 12 cases. Am J Dermatopathol. 2000;22(2):97–103.
62. Leonard N, Smith D, McNamara P. Low-grade malignant eccrine spiradenoma with systemic metastases. Am J Dermatopathol. 2003;25(3):253–255.
63. Jariwala A, Evans A, McLeod G. Not all stubbed toes are innocuous—a case report of rare malignant eccrine spiradenoma (spiradenocarcinoma) of the toe. Foot Ankle Surg. 2010;16(2):e32–e33.
64. Bolognia JL, Jorizzo JL. Adnexal neoplasms. In: Dermatology. 2nd ed. www.expertconsultbook.com. Accessed on January 15, 2011.
65. Lobo I, Amorim I, Selores M. Multiple nodules of the head—clinicopathologic challenge. Multiple eccrine spiradenoma. Int J Dermatol. 2009;48(3):237–238.
66. Andreoli MT, Itani KM. Malignant eccrine spiradenoma: a meta-analysis of reported cases. Am J Surg. 2010 Sep 17. [Epub ahead of print].
67. Ben Brahim E, Sfia M, Tangour M, et al. Malignant eccrine spiradenoma: a new case report. J Cutan Pathol. 2010;37(4): 478–481.
68. Tay JS, Tapen EM, Solari PG. Malignant eccrine spiradenoma. Case report and review of the literature. Am J Clin Oncol. 1997;20(6):552–557.