
James Q. Del Rosso, DO, FAOCD
Dr. Del Rosso is Dermatology Residency Director, Valley Hospital Medical Center, Las Vegas, Nevada. Disclosure: This article was written solely by Dr. Del Rosso, without the assistance or involvement of medical writers. Dr. Del Rosso did not receive compensation for the writing of this article from any company or manufacturer related to any products or formulations mentioned in this article. Dr. Del Rosso serves as a consultant, researcher and/or speaker for Allergan, Coria/Valeant, Galderma, Graceway, Intendis/Bayer, LeoPharma, Medicis, Onset Dermatologics, Ortho Dermatology, Pharmaderm, Promius, Ranbaxy, TriaBeauty, Unilever, and Warner-Chilcott.
Atopic dermatitis (AD) has been described as “the most common itchy and relapsing inflammatory skin condition,” estimated to exhibit a lifetime prevalence of 15 to 30 percent of children and 2 to 10 percent of adults.[1] AD is a common complex disease that is encountered with high frequency and involves several causative genes related to its pathogenesis.[2] AD exhibits polygenic inheritance, with both gene-gene and gene-environment interactions reported to play important pathophysiological roles.[1,2] Familial factors have been shown to markedly affect the likelihood of a given individual being affected clinically by AD. A higher concordance rate has been shown with monozygotic twins as compared to dizygotic twins.[3] Parental history represents the strongest risk factor for the development of AD.[1] In a given individual, the incidence risk is doubled if one parent has had AD and tripled if both parents have a positive history of AD.[1,3]
Additionally, single nucleotide polymorphisms involving genes relevant to immunological responses in AD have also been postulated in the development of this complex disorder.[1]
A notable rise in the incidence of allergic diseases and AD has been observed over time in industrialized countries, with a two- to threefold increase documented over the past three decades.[1] In children, 45 percent of AD cases emerge within the first six months of life, 60 percent of cases emerge within the first 12 months of life, and 85 percent of cases emerge before the age of five years.[1] Some reports suggest a correlation between the markedly increased incidence of allergic disease and AD in industrialized countries with several environmental and extrinsic factors. These include higher levels and density of air pollution, indoor exposure to house dust mites, increased exposure to household pets, widespread use of antibiotics, variations in dietary exposures, and blunted development of immunity in children.[1,4] Interference with complete immunological development in children is believed to be due to decreased or incomplete exposure to bacterial and viral infections as a consequence of improved hygiene, early treatment of infections, and vaccination.[1,4] It has been suggested that exposure to bacteria-derived toxins and/or viral-induced inflammation during childhood promotes maturation of immunity, including Th1 response; however, this hypothesis has not been definitely correlated with the pathogenesis of AD.[1,5,6]
Several pathophysiological factors have been related to AD and are reviewed in detail elsewhere.[1] Among these, epidermal barrier dysfunction inherent to AD has emerged as one of many factors that needs to be addressed when outlining both the initial treatment plan for the acute flare and the long-term management plan for maintenance of remission.[1,7,8] The impaired epidermal barrier associated with AD results in increased transepidermal water loss (TEWL) and reduced water-holding capacity of the stratum corneum, which leads to skin roughness, fine scaling, dryness, microfissuring, heightened susceptibility to penetration of exogenous irritants and allergens, and an increased propensity for skin irritation and pruritus.[7,9] Individuals with AD are affected by the consequences of epidermal barrier dysfunction every day as the aberrations of both their impaired stratum corneum and cutaneous immune response are inherent to AD.[1,7–10] As a result, the daily incorporation of a skin care regimen that enhances epidermal barrier recovery and integrity, minimizes or avoids epidermal barrier dysfunction, maintains skin hydration, does not elicit clinically relevant cutaneous irritation, and has a low propensity to induce allergic contact dermatitis is a vital component of AD management.[8,11] In reality, an optimized daily skin care regimen may contribute to the repair of epidermal barrier dysfunction, sustain epidermal barrier integrity, and serve as an important adjunct to improvement of skin actively affected by AD during an eczematous flare.[8,11–14]
What is meant by “atopic skin”?
The simplest definition of “atopic skin” is skin in individuals correctly diagnosed as having AD. Several publications have defined specific criteria for the diagnosis of AD.[15–18] Most of the criteria designations focus on clinical considerations for diagnosis due to the absence of confirmatory diagnostic tests, with some criteria developed to assist with epidemiological evaluations and population-based studies.[19] Criteria that take into account that anatomic distribution and clinical presentation may differ in the infantile, childhood, and adult phases of AD are significant, especially as many cases of AD in adulthood may present in a more localized fashion. Although properly diagnosed as a form of eczematous dermatitis, clinical presenations in an adult may not be recognized in some cases as an expression of adult AD.18 Examples include hand eczema, foot eczema, nummular eczema, and lichen simplex.
All published diagnostic criteria for AD agree on the following basic features that are hallmarks of AD: 1) pruritus, 2) chronic and/or relapsing disease course, and 3) characteristic morphology and distribution patterns often related to patient age.[15–19] A discussion of the clinical presentations of AD are beyond the scope of this article, but have been reviewed in detail elsewhere.[20–22]
More recently, subtypes of AD have been identified based on the presence or absence of respiratory allergies and immunoglobulin E (IgE) sensitization to aeroallergens and/or food allergens.[19] The mixed type of AD, in addition to the skin manifestations, also exhibits concomitant respiratory allergies, usually allergic rhinitis and/or asthma, and polyvalent IgE sensitization to inhalant and/or food allergens in serum or skin tests. The pure type of AD is not associated with respiratory symptomatology, but may either be positive for polyvalent IgE sensitization to inhalant and/or food allergens (extrinsic AD) or does not exhibit polyvalent IgE sensitization to inhalant and/or food allergens (intrinsic AD). In intrinsic AD, the total serum IgE is not elevated. Overall, extrinsic AD appears to be more common than intrinsic AD, with the latter noted in 10 to 40 percent of cases.[19] The clinical and histological features of intrinsic AD are indistinguishable from those noted in patients with extrinsic AD; however, the onset of intrinsic AD tends to be later, and some patients with intrinsic AD tend to present with a “head-and-neck-type” pattern.[23,24]
As a result of more thorough research defining disease subtypes, a T helper 2 (Th2) cell-related cytokine-release profile is common to both allergic and nonallergic forms of AD and bronchial asthma, thus encompassing both intrinsic and extrinsic AD. It has been suggested that the term “atopy” be narrowed to include only patients with proven IgE-mediated sensitization based on serum or skin testing.[25] However, at present, the term “atopic skin” best defines an inherent predilection and is used to encompass patients with any of the subtypes of AD, either currently active or quiescent. This is an important distinction as chronic and/or relapsing clinical features, such as xerosis, eczematous dermatitis, and pruritus, are common to all subtypes of AD, although patients with loss-of-function mutations of the profilaggrin/filaggrin gene predominantly exhibit extrinsic AD or mixed-type AD.[1,2,13,19]
It has been correctly emphasized that “normal-appearing skin in AD is in fact not normal,” supporting the clinically useful and separate designation of “atopic skin.”[13] The epidermal barrier impairment inherent to AD that is related to innate abnormalities of stratum corneum supports the need for incorporating proper skin care every day as an integral therapeutic component of AD management.
What is known about epidermal barrier function and the stratum corneum in patients with normal skin who do not have a history of “atopic skin” or atopic dermatitis?
The normal stratum corneum (SC) serves as the primary site of the epidermal barrier, exhibiting several physiological and homeostatic functions.[26] The structure of the stratum corneum has been described as “bricks and mortar.” The “bricks” comprise layered corneocytes held together by corneodesmosomes, and the “mortar” comprises parallel lamellar membranes of extracellular physiological lipids present in a defined ratio (intercellular lipid membrane).[26,27]
The protective functions of the SC ascribed to the array of corneocytes (“bricks”) include mechanical shear and impact resistance, initiation of inflammation via cytokine activation, impairment of ultraviolet light penetration, and hydration and water flux.[26] Within corneocytes, the protein filaggrin is vital to SC terminal differentiation and contributes to hydration and water flux through breakdown into several amino acid degradation products, such as urocanic acid (UCA) and pyrrolidone carboxylic acid (PCA).[28–30] The amino acid-derived products of filaggrin breakdown within SC, along with several small compounds including sugars and electrolytes, form natural moisturizing factor (NMF). NMF exhibits powerful humectant properties that serve to sustain SC hydration that is necessary for completion of several important biochemical activities in skin, including proper function of protease enzymes, many involved in the process of normal desquamation of individual corneocytes.[29]
The protective functions of SC ascribed to the intercellular lipid membrane (“mortar”) include permeability barrier; cohesion integrity; desquamation; antimicrobial barrier; exclusion of toxins, irritants, and allergens; selective absorption; and water flux.[26] Precursor extracellular lipids derived from lamellar bodies (LB) present in the granular layer of the SC, namely glycosylceramides, sphingomyelin, and phospholipids, are converted by specific hydrolytic enzymes to ceramides 1–7, ceramides 2 and 5, and free fatty acids, respectively.[26] Collectively, these hydrophobic lipids comprise, along with cholesterol and small quantities of cholesterol sulfate, cholesterol esters and nonpolar lipids, the intercellular lipid membrane (the “mortar” component of the epidermal barrier as described above).[7,10,26] Ceramides account for a family of at least seven subfractions, and as a group comprise approximately 50 percent of SC lipid content by weight.[27] Cholesterol and its derivatives and free fatty acids, account for 25 percent and 10 to 20 percent of SC lipid content by weight, respectively.[27] Importantly, the physiological lipids comprising the intercellular lipid membrane help to sustain SC water content and flux through their relative composition and specialized ompartmentalization in the intercellular spaces in vivo.[31]
The stratum corneum is dynamic with regard to epidermal barrier integrity and repair, with recovery initiated rapidly in response to an insult that increases TEWL by damaging intercellular lipid membrane integrity and composition.[26,27] Factors that decrease NMF, such as improper skin cleansing and use of harsh skin cleansing products, may also precipitate a decrease in SC water content.[8] An increase in TEWL, which leads to skin dessication, quickly triggers an organized metabolic response within the epidermis characterized by enhanced production of SC lamellar lipids, which help to restore barrier function, and an increase in filaggrin production, promotes skin rehydration through increased production of natural moisturizing factor.[26,27]
What is known about epidermal barrier function and the stratum corneum in patients with “atopic skin” and a history of atopic dermatitis?
Several factors associated with AD directly contribute to impaired epidermal barrier function, including reduced SC lipid content, a decrease in some ceramide subfractions in SC, an altered ratio of lipid composition of the SC intercellular lipid membrane, altered activity of some enzymes, and defective filaggrin production, the latter correlating with genetic mutations present in some AD patients.[7,10,13,28,30,32] Hence, epidermal barrier impairment in AD can be due to several mechanisms.
Epidermal water flux in AD. A three- to fivefold increase in TEWL has been demonstrated in lesional skin of patients with AD as compared to visibly uninvolved skin, with the magnitude of barrier impairment correlating with the severity of inflammation of lesional skin.[10,33] Up to a twofold increase in TEWL has been documented in clinically normal skin and in noneczematous xerotic skin of patients with active AD elsewhere, supporting the concept that active AD promotes barrier impairment in both involved or uninvolved skin.[10,33] On the other hand, TEWL and skin hydration assessments in normal-appearing skin of AD patients free of exacerbations for more than five years were not different from normal controls.[10]
Stratum corneum lipids in AD. Alteration in SC lipids appears to markedly contribute to epidermal barrier dysfunction in AD.[7,10,31,34–36] A significant reduction in total skin lipids from nonlichenified forearm skin of subjects with AD has been noted as compared to controls with normal skin or ichthyosis vulgaris without concomitant AD.[7,10] The decreased epidermal lipids in AD that have been reported include multiple ceramide subfractions, phospholipids, and sterol esters, with an increase in free fatty acids and sterols also identified.10 Importantly, it has been noted that unlike several ceramide subfractions, the relative levels of several SC lipid classes, including free fatty acids, triglycerides, cholesterol, cholesterol esters, cholesterol sulfate, squalene, and phospholipids, are similar in skin from subjects with AD as compared to controls, while some studies have shown some differences between groups in some individual lipid classes.[7,10,36] More recently, it is believed that a previously reported decrease in levels of phospholipids may actually reflect a decrease primarily in sphingomyelin, a precursor of ceramide production.[10,26] Other investigators have documented an incomplete transformation of epidermal phospholipids into other lipid classes in subjects with AD.[10,35]
The major lipid subclass associated with a notable decrease in SC content in subjects with AD as compared to the SC of control groups is ceramides.[7,10,31,34,36] A marked decrease in SC ceramides has been shown in clinically uninvolved plantar skin of patients with AD versus controls, in back skin and toenails of patients with AD, and in lesional forearm skin and nonlesional skin in AD patients.10 Additionally, a significant reduction of ceramides has been documented in lesional forearm SC of patients with AD, with marked ceramide reductions also noted in nonlesional SC from patients affected with AD.[10,31,34] Among the ceramide subfractions, the greatest decrease in AD subjects has been noted with ceramide 1 in both lesional and nonlesional skin; however, ceramides 2 through 6 were also markedly reduced in both lesional and nonlesional skin.[7,10,31,34] In subjects with AD, a reduction in ceramide 1 and in total ceramide content was also found in SC of clinically dry skin that was otherwise devoid of clinical signs of eczematous dermatitis, that is, “atopic dry skin.”[10,36,37] Correlation of SC ceramide content with epidermal barrier function in subjects with AD demonstrated a marked decrease in the amounts of ceramide 1 and ceramide 3, with an increase in TEWL correlating significantly with the reduction in ceramide 3.[7,10,38] Studies completed with different inhibitors of ceramide synthesis demonstrate “a broad requirement for the ceramide family in barrier function.”[10]
Ultimately, the changes in SC lipid fractions observed in AD, as described previously, leads to alteration of the relative ratio of SC lipids. The resultant effect in patients with AD is a fundamental disturbance in the balance of SC lipid composition in both lesional and nonlesional skin.
Stratum corneum enzyme activity in AD. In normal skin, regulation of SC ceramide balance involves the interplay between four major enzymes, beta-glucocerebrosidase, acid sphingomyelinase, ceramidase, and serine palitoyl transferase.[10,39] Evaluation of beta-glucocerebrosidase and ceramidase activities in SC in nonlesional (noneczematous) dry skin from subjects with AD versus age-matched controls showed no differences in activity.[10,40] Direct assays of acid sphingomyelinase activity in SC of AD patients has not been completed; however, content measurement of enzyme protein demonstrated a slightly increased amount of acid sphingomyelinase in lesional as compared to nonlesional skin, possibly suggesting minimalization of its role in AD pathogenesis.[10] Propasin—a precursor protein of sphingolipid activator proteins which promote hydrolysis of sphingolipids, such as glucosylceramides and sphingomyelin—appears to be required for normal epidermal barrier formation.[10,41] Decreased propasin levels have been observed in the epidermis of AD subjects, and may be associated with diminished activation of beta-glucocerebrosidase acid and sphingomyelinase in AD, potentially modifying ceramide formation and balance via modulation of enzyme function.[10,42]
Decreased SC ceramide production in AD patients has also been associated with the observation of an abnormal expression of a sphingomyelin deacylase (SMD) enzyme, glucosylceramide/ sphingomyelin deacylase, in both lesional and nonlesional SC.[10,13,43] An eight-fold increase of SMD activity in lesional skin, and a five-fold increase in SMD activity in nonlesional skin, has been documented.[10,44]
As a result of this increased activity of SMD in AD, sphingomyelin hydrolysis is significantly elevated in both lesional and nonlesional SC of subjects with AD, thus circumventing ceramide production. Rather, SMD promotes the release of free fatty acids and sphingosylphosphoryl-choline, both of which may serve as lipid signals involved in the pathogenesis of AD.[10] The discovery of abnormal expression and increased activity of SMD in AD suggests that decrease in SC ceramide content and barrier impairment are at least partially related to altered and accelerated sphingomyelin metabolism.[10,43,44]
Filaggrin production in stratum corneum in AD. Proteins involved in normal epidermal differentiation, such as filaggrin and involucrin, are vital components of epidermal integrity, including barrier function. As stated above, filaggrin breakdown in SC leads to formation of the amino acid-derived components of NMF, while involucrin, a cornified envelope (CE) protein, serves as a substrate for the covalent binding of ceramides to the CE.[7,10,27,28,30,37] Reduced expression of both filaggrin and involucrin has been noted in lesional skin of AD, while reduced filaggrin expression was also shown in nonlesional skin.[10,45]
The relatively recent finding of loss-of-function mutations of the profilaggrin/filaggrin gene is a major discovery related to risk for development of ichthyosis vulgaris and AD, the latter often in combination with true sensitization (elevated IgE levels) and development of asthma and possibly allergic rhinitis.[1,22,28,30] Subjects heterozygous for filaggrin gene mutations exhibited mild ichthyosis vulgaris and AD, while those with homozygous or compound heterozygous mutations in filaggrin genes all presented with AD and with severe ichthyosis vulgaris.[22,28,30] Reduced expression of filaggrin leads to overall impairment in epidermal barrier function in multiple ways. Importantly, these include 1) the reduced ability to form the homeostatic quantities of NMF required for normal physiological barrier function and water flux and 2) decreased accessibility for increased filaggrin expression as a “barrier repair response” after insult from external factors which impair barrier integrity and function.
Other currently unknown genetic variations in epidermal components are also likely to be involved in the pathogenesis of AD, especially those localized in the epidermal differential complex (EDC) on chromosome 1q21.1 It has been suggested that genetic factors affecting stratum corneum chymotryptic enzyme (SCCE) may play a role in AD and epidermal barrier impairment.[46] Further research is welcome in this exciting area of genetic research and the pathogenesis of AD.
How does impairment of epidermal barrier function correlate with the pathogenesis of atopic dermatitis?
Inflammation associated with inflammatory cutaneous disorders, such as AD, rosacea, and psoriasis, is often also associated with epidermal barrier impairment, which is often expressed as an increase in TEWL.[8,26,27] Based on our current knowledge, it is difficult to clearly determine if epidermal barrier impairment plays a primary initiating role in the pathogenesis of AD or is a secondary consequence of cutaneous inflammation, and there are arguments to support both views.[10] This author believes that epidermal (and mucosal) barrier impairment both contribute to the pathogenesis of AD and occurs secondary to and/or is exacerbated by eczematous inflammation. As suggested by others, a combination of two distinct major features of AD, immunological abnormalities and mucocutaneous barrier dysfunction, work in concert to precipitate the clinical and laboratory manifestations of AD.[10,47] The presence of epidermal barrier impairment in clinically normal skin of individuals with AD as compared to controls also supports a primary role in the pathogenesis of AD.[7,10,32,33,36,38]
As a consequence of epidermal barrier impairment, the skin of AD patients is believed to be more susceptible to penetration by haptens as well as high molecular weight structures or particles, such as allergens, bacteria, and viruses.[1,10] It has been suggested that the increased incidence of AD in industrialized countries relates to several domestication and lifestyle factors, which 1) may promote epidermal barrier degradation, such as increased washing/bathing frequency, greater use of soaps and shampoos, and common exposure to forced dry-air heating; 2) may increase overall exposure to aeroallergens, such as pollens and house dust mites via air conditioning and poor indoor ventilation; and 3) may increase earlier systemic exposure to potential allergens via feeding of animal proteins to infants.10 Increased and repeated exposures to multiple allergens from many sources, enhanced by greater susceptibility to skin penetration, promotes “nonspecific hypersensitivity,” induction and perpetuation of inflammation, and further degradation or worsening of barrier function.[10,47] These events repetitively cycle as if on a “hamster wheel,” explaining the chronicity of AD, with common persistence into adulthood presenting as less obvious forms of AD. These adult presentations include “atopic” dry skin (xerosis), pruritus, poor skin accommodation to topical irritants or low ambient humidity with precipitation of clinical symptoms and/or signs of eczema, and localized forms of eczema, such as hand eczema, foot eczema, recurrent eyelid dermatitis, and lichen simplex.[10,18,48]
Another important correlation with permeability of the epidermal barrier that may indirectly support a primary relationship between epidermal barrier compromise and the pathogenesis of AD is dendritic cell (DC) density. Langerhans cells (LCs) play a major role in antigen recognition and the initiation of allergic immune response and also prime the conversion of naïve T lymphocytes into Th2-type lymphocytes; however, LCs do not typically exhibit a strong innate proinflammatory profile.1 Murine studies in which the epidermal barrier is compromised by topical agents known to degrade SC barrier function, or through tape stripping, demonstrate increased LC density and augmented expression of some antigens by subsets of LCs.[10,49] Thus, it appears that epidermal barrier disruption can increase the potential for allergic contact dermatitis through enhanced penetration of allergens or haptens and through modification of immune response which can augment activation of T lymphocytes.10 Additionally, the epidermis of subjects with AD and IgE sensitization contains LCs, which express the high-affinity receptor for IgE, and also another subset of migratory DC called inflammatory dendritic epidermal cells (IDECs), which highly express the high-affinity receptor for IgE in AD.[1,50] Unlike LCs, high-affinity IgE-receptor-positive IDECs amplify allergic immune response through marked production of proinflammatory cytokines and appear to facilitate the biphasic “Th2-Th1 switch” that occurs with transition of acute to chronic AD lesions.[1,51]
The hyper-reactive nature of skin in AD is not only affected by allergen exposure, but also through contact with topical irritants.[10] In addition to lesional skin, an abnormal SC is also present in clinically uninvolved skin in AD, with an increase in TEWL observed for one week after application of a topical irritant.[13] Observations after irritant skin exposure in subsets of AD subjects as compared to other tested groups suggest that epidermal barrier dysfunction may play an important role in the elicitation of AD, even in the absence of cutaneous allergen exposure. It has been shown that subjects with a history of AD exhibited higher TEWL measurements after irritant exposure as compared to those with a history of only allergic contact dermatitis or normal controls and also demonstrated greater susceptibility to irritants in clinically xerotic skin as compared to visibly normal skin.[10,52] Both atopic individuals without a history of AD, and those with a history of AD but without currently active eczematous skin lesions, exhibit an increased response to challenge with a topical irritant.[10] Additionally, it has been noted that patients with AD demonstrate a greater magnitude of both damage to the epidermal barrier and inflammation after topical irritant exposure as compared to those with only allergic seasonal rhinitis and no history of AD, including when the seasonal rhinitis is symptomatic or quiescent.[10,53] Ultimately, both specific (allergenic) and nonspecific (irritant) triggers elicit cutaneous inflammation and contribute to the chronicity of AD.[13]
Although explanations supporting a correlation between epidermal barrier function and the pathogenesis of AD are reviewed here, more detailed discussions are reviewed elsewhere.[10,47] Nevertheless, there appears to be an innate epidermal barrier impairment that is continually present in individuals with AD, is worsened in actively eczematous skin during a flare of AD, and remains present in both normal and xerotic-appearing skin when these individuals with AD return to a period of remission.
What skin care approaches have been shown to improve atopic dermatitis?
The use of gentle cleansers incorporating synthetic detergents (“syndets”) with minimal irritation potential and moisturizers containing a variety of humectants, occlusive agents, and lipids have been used in the management of xerosis and AD.[8,13,14] Some studies addressing skin moisturization and barrier repair therapy report adjunctive clinical benefit in the treatment of active AD, with a few trials, although more limited, noting prolongation of remission between flares and reduction in usage of topical corticosteroid therapy.[7,8,10,13,14,26,27,29,31,54] It would not be accurate to state that there is one single approach to fundamental skin care in AD or that one or a few products have been definitively shown to be superior to all others. Nevertheless, it is important that poorly formulated or non-gentle cleansers be avoided, such as soap-based agents, those which contain additives that are likely irritants or reasonably common allergens, and those that contain “special additives” that may be abrasive or irritant in nature.[8,13] Gentle cleansers capable of depositing lipid into the epidermis are likely to be advantageous.[29] Moisturizers vary in composition, and have classically incorporated occlusive agents to more expeditiously reduce TEWL, and humectants to promote and sustain SC hydration.[8,13,26] More recently, several moisturizers incorporate as a part of their overall formulation a variety of lipids in an attempt to restore SC lipids that have been lost after insult to the epidermis. Some of these lipids contained in moisturizers may be actively incorporated into lamellar bodies.[10,13,26,29,31] The specific characteristics, quantities, and relative ratios of the lipids used in individual moisturizers may directly influence the time course and substantivity of barrier repair and function.[26]
Many formulations, including both over-the-counter (OTC) moisturizers and “barrier repair” creams available by prescriptions, incorporate various ceramide subfractions, ceramide precursors, and/or pseudoceramides, based on the plethora of data demonstrating a decrease in ceramide content in SC of AD patients.[7,10,13,31,54–58] A review of all these formulations and the data supporting their clinical use is beyond the scope of this article. Importantly, the clinician must consider the therapeutic merits and possible disadvantages of a variety of cleansers and moisturizers based on available data, clinical experience, and patient-specific factors. The next section of this article discusses the components of a body wash and moisturizer product regimen targeted specifically for use in “atopic skin.”
What are the components of the skin care regimen targeted for use in atopic skin that incorporates both a body wash and moisturizer?
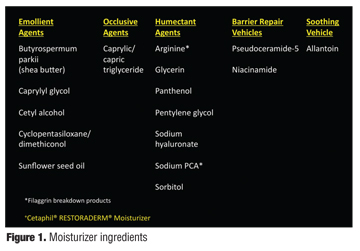
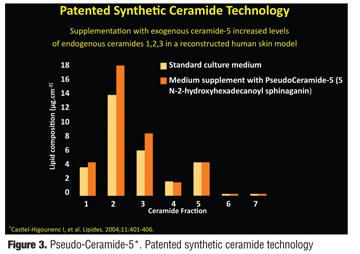
A skin care product combination, which includes both a body wash (Cetaphil Restoraderm Body Wash, Galderma Laboratories, Fort Worth, Texas) and moisturizer (Cetaphil Restoraderm Lotion, Galderma Laboratories) has been developed with the recommendation that this product combination be used for “atopic skin.”[58,59] The components of the moisturizer (CRM) and body wash (CRBW) in this product combination are categorized and listed in Figure 1 and Figure 2, respectively.[58] The moisturizer includes occlusive, emollient, and humectant agents needed to formulate an effective moisturizer by decreasing evaporative water loss, enhancing epidermal hydration by attracting water flux upward from the dermis into the SC, and augmenting ease of spread and occlusivity, respectively.[8,29] Also included in the moisturizer are the filaggrin breakdown products, sodium PCA and arginine, unique to this product line, and pseudoceramide-5 [N-(2-hydroxyhexadecanoyl) sphinganin], a patented exogenous ceramide precursor.[58,60] The application of pseudoceramides in combination with other intercellular lipids in lipid-depleted stratum corneum has been shown to markedly increase SC bound-water content.[31] Various pseudoceramides have been shown to exert water-holding properties in SC similar to those seen with natural ceramides, and clinical trials evaluating pseudoceramide-containing creams have demonstrated their clinical efficacy in the management of xerosis, including cases observed in AD patients.[31] An in-vitro study using a reconstructed human skin model demonstrated that the addition of pseudoceramide-5 to the medium resulted in an increase in ceramides 1, 2, and 3 (Figure 3).[60] The investigators, through use of specific assays, excluded both the incorporation of exogenous pseudoceramide-5 into the SC intercellular lipid membrane and the induction of lipogenesis as reasons for the increase in ceramide subfractions that were documented after the addition of pseudoceramide-5. Rather, they claim that the increase in ceramides 1, 2, and 3 demonstrate that the exogenous pseudoceramide-5 was transformed into endogenous ceramides.[60]
{kind=link}
{kind=link}
{kind=link}
Other major components of CRM and CRBW include sunflower seed oil and shea butter, with panthenol also present in the CRM.[58] Sunflower seed oil (Helianthus annuus) contains lipids similar in composition to SC lipids and has been shown after topical application to markedly increase ceramide synthesis and also cholesterol synthesis.[61,62] Shea butter (Butyrospermum parkii) is derived from the Shea tree fruit; contains stearic acid, linoleic acid, and catechins (antioxidants); is commonly found in topical formulations used for inflammatory dermatoses such as AD and psoriasis; and does not appear to exhibit allergenicity after topical or systemic exposure.[63-65] Niacinamide (viamin B3) has been shown in vitro to increase the synthesis of glucosylceramides by 7.4-fold and sphingomyelin by 3.1-fold, both of which are ceramide precursors.[66] A 2.3-fold increase in free fatty acids and a 1.5-fold increase in cholesterol were also noted.[66] Topical application of niacinamide to subjects with xerotic skin demonstrated an increase in ceramides that correlated directly with reduction in TEWL.[66] Panthenol has been shown to assist epidermal barrier function by reducing TEWL and increasing SC hydration, with noted utility in AD and other skin disorders associated with epidermal barrier impairment.[67,68] A variety of clinical studies have been performed in individuals with atopic skin including infants and toddlers with atopic skin. The results of these studies are expected to be published soon.
What final conclusion can be drawn from available information on epidermal barrier dysfunction in atopic dermatitis and therapeutic measures taken using skin care as a component of management?
The information discussed in this paper focuses on the relationship between epidermal barrier impairment and AD, including a discussion of “atopic skin.” Importantly, in addition to SC aberrations and altered barrier function, a large body of evidence has implicated a variety of diverse changes and mechanisms in the pathogenesis of AD, including genetics, environmental factors, altered immunological responses, changes in immune cell types, neurogenic factors, and decreased cutaneous antimicrobial defense.[1,10] Therefore, AD is a complex disease, characterized by variable severity and chronicity. As onset is usually in early childhood, measures to reduce the severity of AD, including frequency and intensity of intermittent flares, is of utmost importance. Unfortunately, most studies on therapy of AD evaluate the response of acute flares to active medical therapy (i.e., topical corticosteroid, topical calcineurin inhibitor), with few long-term studies available on management of AD.
At the present time, based on information we have relating epidermal barrier impairment and AD, it seems prudent to utilize a proper daily skin care regimen in patients with AD during periods of both inactive and active disease. At minimum, we know that proper skin care can assist in maintaining epidermal barrier function, decrease signs and symptoms of xerosis, and improve the overall quality of skin. Although studies are limited, proper skin care directed at moisturization and barrier repair may potentially reduce the frequency and/or intensity of flares and/or decrease the overall use of topical medications. Certainly, more studies are needed on long-term AD management, including the impact of skin care regimens. Nevertheless, we must work with our best clinical judgment and the information we have in hand, with gentle cleanser use and moisturization felt to be an important component of AD management.[7,8,10,13,14,26,27,29,54]
Product selection is aimed at choosing a cleanser that is nonirritating, does not contribute to barrier compromise, and may assist in skin hydration depending on the components and nature of the formulation. Moisturizer selection is aimed at products that are not irritating, not likely to exhibit allergenicity, and are capable of optimizing skin hydration and barrier function in AD. More recently, moisturizer technology has focused on SC lipid replenishment in addition to providing humectant and occlusive qualities. This is especially true with the advent of moisturizers containing ceramides, ceramide precursors, and/or pseudoceramides. A more recently introduced skin care product combination, inclusive of a body wash (CRBW) and moisturizer (CRM), is reviewed in this paper. The formulation design of both the body wash and moisturizer has been positioned for use as daily skin care in atopic skin, incorporating a combination of ingredients, such as humectants (including filaggrin degradation products), occlusive/emollients, and barrier repair agents (pseudoceramide-5 and niacinamide). The components of the formulation suggest that this skin care product combination appears to be a viable option for patients with atopic skin.
References
1. Bieber T, Prolss J. Atopic dermatitis. In: Gaspari AA, Tyring SK, eds. Clinical and Basic Immunodermatology. London: Springer-Verlag; 2008:193–206.
2. Elliott K, Forrest S. Genetic of atopic dermatitis. In: Bieber T, Leung DYM, eds. Atopic Dermatitis. New York: Marcel Dekker; 2002:81–110.
3. Schultz Larsen FV, Holm NV. Atopic dermatitis in a population based twin series: concordance rates and heritability estimation. Acta Derm Venereol (Stockh). 1985;114(Suppl):159.
4. Strachan DP. Hay fever, hygiene, and household size. Br Med J. 1989;299:1259–1260.
5. Zutavern A, Hirsch T, Leupold W, et al. Atopic dermatitis, extrinsic atopic dermatitis and the hygiene hypothesis: results from a cross-sectional study. Clin Exp Allergy. 2005;35:1301–1308.
6. Williams H, Flohr C. How epidemiology has challenged three prevailing concepts about atopic dermatitis. J Allergy Clin Immunol. 2006;118:209–213.
7. Di Nardo A, Wetz PW. Atopic Dermatitis. In: Leyden JJ, Rawlings AV, eds. Skin Moisturization. 1st ed. New York: Marcel Dekker; 2002:165–178.
8. Del Rosso JQ. Understanding skin cleansers and moisturizers: the correlation of formulation science with the art of clinical use. Cosmet Dermatol. 2003;16:19–31.
9. Thune P. Evaluation of the hydration and water-holding capacity in atopic skin and so-called dry skin. Acta Derm Venereol. 1989;144 (Suppl):133–135.
10. Proksch E, Elias PM. Epidermal barrier in atopic dermatitis. In: Bieber T, Leung DYM, eds. Atopic Dermatitis. New York: Marcel Dekker; 2002:123–143.
11. Tabata N, O’Goshi K, Zhen YX, et al. Biophysical assessment of persistent effects of moisturizers after their daily applications: evaluation of corneotherapy. Dermatology. 2000;200:308–313.
12. Hanifin JM, Hebert AA, Mays SR, et al. Effects of a low-potency corticosteroid formulation plus a moisturizing regimen in the treatment of atopic dermatitis. Curr Therapeutic Res. 1998;59:227–233.
13. Boguniewicz M. Conventional topical treatment of atopic dermatitis. In: Bieber T, Leung DYM, eds. Atopic Dermatitis. New York: Marcel Dekker; 2002:453–477.
14. Lucky AW, Leach AD, Laskarzewski P, et al. Use of an emollient as a steroid sparing agent in the treatment of mild to moderate atopic dermatitis in children. Pediatr Dermatol. 1997;14:321–324.
15. Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol (Stockh). 1980;92(Suppl):44–47.
16. Diepgen TL, Fartsch M, Hornstein OP. Evaluation and relevance of atopic basic and minor features in patients with atopic dermatitis and in the general population. Acta Derm Venereol (Stockh). 1989;144:50–54.
17. Williams HC, Pembrokle AC, Burney PGF, et al. Community validation of the UK Working Party’s diagnostic criteria for atopic dermatitis. Br J Dermatol. 1996;135:12–17.
18. Schultz Larsen F, Diepgen T, Svensonn A. Clinical criteria in diagnosing AD: the Lillehammer criteria 1994. Acta Derm Venereol (Stockh). 1996;76(Suppl 196):115–119.
19. Wuthrich B, Schmid-Grendelmeier P. Definition and diagnosis of intrinsic versus extrinsic atopic dermatitis. Conventional topical treatment of atopic dermatitis. In: Bieber T, Leung DYM, eds. Atopic Dermatitis. New York: Marcel Dekker; 2002:1–20.
20. Paller AS, Mancini AJ. Exczematous eruptions in childhood. In: Paller AS, Mancini AJ, eds. Hurwitz Clinical Pediatric Dermatology, 3rd Edition. Philadelphia: Elsevier-Saunders; 2006:49–65.
21. Krafchik BR, Halbert A, Yamamoto K, Sasaki R. Atopic dermatitis. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology, 3rd Edition. Philadelphia: Mosby-Elsevier; 609–629.
22. Mallory SB, Bree A, Chern P, Eczematous dermatoses. In: Mallory SB, Bree A, Chern P, eds. Illustrated Manual of Pediatric Dermatology. Boca Raton, FL: Taylor & Francis; 2005:49–57.
23. Werfel T, Kapp A. What do we know about the epidemiology of the intrinsic type of atopic dermatitis? Curr Probl Dermatol. 1999;28:29–36.
24. Jensen-Jarolim E, Poulsen LK, With H, et al. Atopic dermatitis of the face, scalp, and neck: type I reaction to the yeast Pityrosporum ovale? J Allergy Clin Immunol. 1992;89:44–51.
25. Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol. 2004;113:832–836.
26. Elias PM. Physiologic lipids for barrier repair in dermatology. In: Draelos ZD, ed. Cosmeceuticals. 1st ed. Philadelphia: Elsevier-Saunders; 2005; 63–70.
27. Harding CR. The stratum corneum: structure and function in health and disease. Derm Ther. 2004;17:6–15.
28. O’Regan GM, Sandilands A, McLean WHI, Irvine AD. J Allergy Clin Immunol. 2008;122:689–693.
29. Rawlings AV, Canestrari DA, Dobkowski B. Moisturizer technology versus clinical performance. Derm Ther. 2004;17:49–56.
30. McLean I. Loss-of-function mutations in the filaggrin gene lead to reduced level of natural moisturizing factor in the stratum corneum. J Invest Dermatol. 2008;128:2117–2119.
31. Imokawa G. Ceramides as natural moisturizing factors and their efficacy in dry skin. In: Leyden JJ, Rawlings AV, eds. Skin Moisturization. 1st ed. New York: Marcel Dekker; 2002:267–302.
32. Cook M, Danby S, Vasilopoulos Y, et al. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol. 2009;129:1892–1908.
33. Seidenari S, Giusti G. Objective measurement of the skin of children affected by atopic dermatitis: a study of pH, capacitance and TEWL in eczematous and clinically uninvolved skin. Acta Derm Venereol. 1995;75:429–433.
34. Imokawa G, Abe A, Higaki Y, et al. Decreased level of ceramides in stratum corneum of atopic dermatitis: an etiologic factor in atopic dry skin? J Invest Dermatol. 1991;96:523–526.
35. Schafer L, Kragballe K. Abnormalities in epidermal lipid metabolism in 11 patients with atopic dermatitis. J Invest Dermatol. 1991;96:10–15.
36. Yamamoto A, Serizawa S, Ito M, et al. Stratum corneum abnormalities in atopic dermatitis. Arch Dermatol Res. 1991;283:219–223.
37. Matsumoto Y, Hamashima H, Masuda K, et al. The antibacterial activity of plaunotol against Staphylococcus aureus isolated from the skin of patients with atopic dermatitis. Microbios. 1998;96:149–155.
38. DiNardo A, Wertz P, Gianetti A, et al. Ceramide and cholesterol composition of the skin of patients with atopic dermatitis. Acta Derm Venereol. 1998;78:27–30.
39. Choi MJ, Maibach HI. Role of ceramides in barrier function of healthy and diseased skin. Am J Clin Dermatol. 2005;6:215–223.
40. Jin K, Higaki Y, Takagi Y, et al. Analysis of beta-clucocerebrosidase and ceramidase activities in atopic and aged dry skin. Acta Derm Venereol. 1994;74:337–340.
41. Doering T, Holleran WM, Potratz A, et al. Sphingolipid activator proteins are required for epidermal permeability barrier formation. J Biol Chem. 1999;274:11038–11045.
42. Cui CY, Kusuda S, Seguchi T, et al. Decreased level of propasin in atopic skin. J Invest Dermatol. 1997;109:319–323.
43. Hara J, Higuchi K, Okamoto R, et al. High-expression of sphingomyelin deacylase is an important determinant ceramide deficiency leading to barrier disruption in atopic dermatitis. J Invest Dermatol. 2000;115:406–413.
44. Murata Y, Ogata J, Higaki Y, et al. Abnormal expression of sphingomyelin acylase in atopic dermatitis: an etiologic factor for ceramide deficiency? J Invest Dermatol. 1996;106:1242–1249.
45. Seguchi T, Cui CY, Kusuda S, et al. Decreased expression of filaggrin in in atopic skin. Arch Dermatol Res. 1996;288:442–446.
46. Vasilopoulos Y, Cork MJ, Murphy R, et al. Genetic association between AACC insertion in the 3’UTR of the stratum corneum chymotryptic enzyme gene and atopic dermatitis. J Invest Dermatol. 2004;123:62–66.
47. Ogawa H, Yoshiike TJ. A speculative view of atopic dermatitis: barrier dysfunction in pathogenesis. Dermatol Sci. 1993;5:197–204.
48. Mar A, Tarn M, Jolley D, et al. The cumulative incidence of atopic dermatitis in the first 12 months among Chinese, Vietnamese, and Caucasians infants born in Melbourne, Australia. J Am Acad Dermatol. 1999;40:597–602.
49. Proksch E, Brasch J. Influence of epidermal permeability barrier disruption and Langerhans cell density on allergic contact dermatitis. Acta Derm Venereol. 1997;77:102–104.
50. Wollenberg A, Kraft S, Hanau D, et al. Immunomorphological and ultrastructural characterization of Langerhan cells and a novel, inflammatory dendritic epidermal cell (IDEC) population in lesional skin of atopic eczema. J Invest Dermatol. 1996;106:446–453.
51. Novak N, Bieber T. The role of dendritic cell subtypes in the pathophysiology of atopic dermatitis. J Am Acad Dermatol. 2005;53(Suppl 2):S171–S176.
52. Tupker RA, Pinnagoda J, Coenraads PJ, et al. Susceptibility to irritants: role of barrier function, skin dryness, and history of atopic dermatitis. Br J Dermatol. 1990;123:199–205.
53. Conti A, Di Nardo A, Seidenari S. No alteration of biophysical parameters in the skin of subjects with respiratory atopy. Dermatology. 1996;192:317–320.
54. Chamlin SL, Kao J, Frieden IJ, et al. Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: changes in barrier function provide a sensitive indicator of disease activity. J Am Acad Dermatol. 2002;47:198–208.
55. Cerave Hydrating Cleanser, Cream, Lotion. Madison, NJ: Coria Laboratories/Valeant Pharmaceuticals; data on file.
56. Epiceram Topical Emulsion [package insert]. Bridgewater, NJ: Promius; 2010.
57. Hyalotopic Plus [package insert]. Cumberland, RI: Onset Dermatologics; 2010.
58. Cetaphil Restoraderm Moisturizer. Fort Worth, TX: Galderma Laboratories; data on file.
59. Cetaphil Restoraderm Body Wash. Fort Worth, TX: Galderma Laboratories; data on file.
60. Castiel-Higounenc I, Chopard M, Ferraris C. Stratum corneum lipids: specificity, role, deficiencies and modulation. Oleagineux, Corps Gras, Lipides. 2004;11:401–406.
61. Eichenfield LF, McCollum A, Msika P. The benefits of sunflower oleodistillate (SOD) in pediatric dermatology. Pediatr Dermatol. 2009;26:669–675.
62. Piccardi N, Piccirilli A, Choulot JC, et al. Sunflower oil oleodistillate for atopy treatment: an in-vitro and clinical investigation. J Invest Dermatol. 2001;117:390–423.
63. Fowler J , Silverburg N. Active naturals have a key role in atopic dermatitis. Semin Cutan Med Surg. 2008;27(Suppl 3):8–10.
64. Maranz S, Wiesman Z, Garti N. Phenolic constituents of Shea (Vitellaria paradoxa) kernels. J Agric Food Chem. 2003;51:6268–6273.
65. Obeda M, Sousselier L. Shea butter: the revival of an African wonder. Global Cosmetic Ind. 1999;4:36–41.
66. Tanno O, Ota Y, Kitamura N, et al. Nicotinamide increases biosynthesis of ceramides as well as other stratum corneum lipids to improve the epidermal permeability barrier. Br J Dermatol. 2000;143:524–531.
67. Ghering W, Gloor M. Effect of topically applied dexpanthenol on epidermal barrier function and stratum corneum hydration. Arzneim-Forsch/Drug Res. 2000;50:659–663.
68. Ebner F, Heller A, Rippke F, et al. Topical use of dexpanthenol in skin disorders. Am J Clin Dermatol. 2002;3:427–433.