
 J Clin Aesthet Dermatol. 2019;12(5):49–53
J Clin Aesthet Dermatol. 2019;12(5):49–53
This ongoing column explores emerging treatment options, drug development trends, and pathophysiologic concepts in the field of dermatology.
by James Q. Del Rosso, DO
Dr. Del Rosso is Research Director of JDR Dermatology Research in Las Vegas, Nevada; is with Thomas Dermatology in Las Vegas, Nevada; and is Adjunct Clinical Professor (Dermatology) with Touro University Nevada in Henderson, Nevada.
FUNDING: There was no funding related to the development, writing, or publication of this article.
DISCLOSURES: Dr. Del Rosso is a consultant, researcher and/or speaker related to this subject area, for Almirall, AnaptysBio, Botanix, Dermavant, Dermira, Galderma, LaRoche Posay, Loreal, Leo Pharma, Pfizer, Ralexar, Regeneron, Sanofi-Genzyme, Valeant.
ABSTRACT: Many systemic therapies are under development for atopic dermatitis, many of which are injectable monoclonal antibodies that inhibit specific pathways involved in the pathogenesis of the disease. Most are currently under development, however, there are preliminary studies with subcutaneous omalizumab (IgE-directed therapy), and oral apremilast (phosphodiesterase-4 [PDE-4] inhibition. Further studies are needed with these agents. This article discusses various agents that address several potential therapeutic approaches, including IgE-directed therapy, anti-IL-31, anti-IL-5, anti-IL-22, PDE-4 inhibition, and thymic stromal lymphopoietin (TSLP)-directed therapy.
KEYWORDS: Atopic dermatitis, targeted therapy, Immunoglobulin E-directed therapy, anti-IL-31, anti-IL-5, anti-IL-22, phosphodiesterase-4, TSLP
In the inaugural article of the present “What’s New” section of this journal, published in February 2019, the management of atopic dermatitis (AD) was discussed. Special emphasis was placed on systemic therapies for cases of AD that are not well controlled with topical therapy and occasional systemic corticosteroid use.1 The major focus of this first article was a discussion of monoclonal antibody therapies that mitigate the effects of overexpression of interleukin-4 (IL-4) and/or IL-13, both of which are reported to be major contributing factors in the pathophysiology of AD.2 There are several pathophysiologic “circuits” that might be operative to varying magnitudes in people with AD, which explains, at least to some degree, the variability in phenotypic expressions of AD and other atopic disorders.2–8 More recent research on systemic treatments for AD, primarily for moderate-to-severe disease that is poorly responsive to conventional therapies, includes various agents that fall under conveniently described categories. These categories are based on specific modes of action (MOA) that antagonize the proinflammatory effects induced by one or more of the following: immunoglobulin E (IgE), IL-4, IL-13, IL-5, IL-6, IL-31, IL-22, IL-12/23, IL-17, or thymic stromal lymphopoietin (TSLP); via inhibition of Janus kinase (JAK) receptors or phosphodiesterase-4 (PDE-4); via antagonism of histamine-4 receptors; or through modulation of aryl hydrocarbon receptors.9–12 Importantly, these categories are not mutually exclusive, in that MOA can overlap in some cases regarding the molecular and biologic effects that any given agent might induce to produce a therapeutic effect.
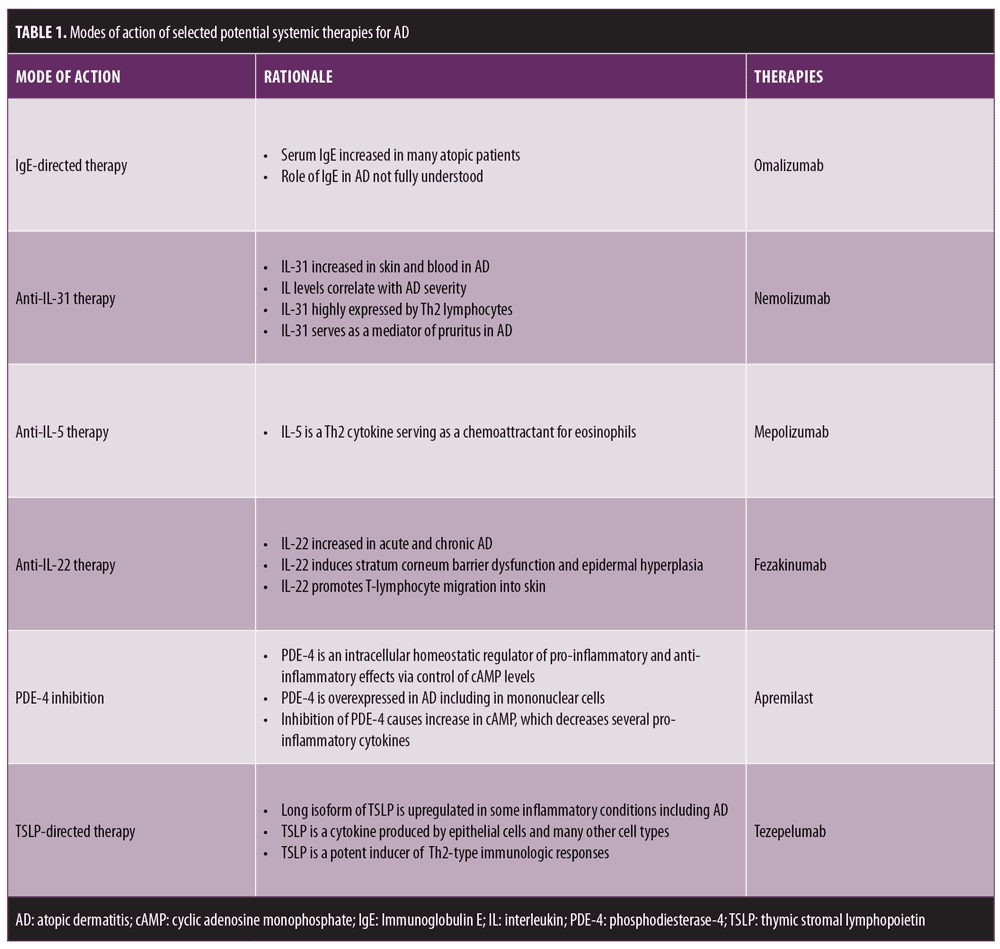
This article discusses some of these therapeutic categories and the currently available data on the systemic agents that fall under selected drug classes (Table 1). With the exception of omalizumab (IgE-directed) and apremilast (PDE-4-directed), all of the therapies reviewed here are under experimental investigation and are not currently available for any indication. As noted above, anti-IL-4/IL-13 (dupilumab) and anti-IL-13 (lebrikizumab, tralokinumab) strategies were discussed in a recent previous article.1 JAK inhibitors, anti-IL-12/23 therapies, and anti-IL-17 therapies will be discussed in future articles.
IgE-Directed Therapy
Although IgE is often thought of as pivotal within the spectrum of atopic diseases, the role of IgE in the pathophysiology of AD is not clear.10,13 Omalizumab, which is approved by the United States Food and Drug Administration (FDA) for the treatment of refractory asthma and chronic idiopathic urticaria, is a subcutaneously administered, recombinant, humanized monoclonal antibody that binds to free or B-cell membrane-bound IgE and reduces free serum IgE levels, resulting in the inhibition of IgE binding to its receptors.9 As a result of binding with IgE, omalizumab reduces both immediate and late-phase allergic inflammation.
Thus far, results achieved with omalizumab for the treatment of AD have yielded conflicting results, with a broad range of therapeutic responses observed; some studies and case reports have documented improvements of AD, while others showed little to no efficacy.9–11,15–17 It has been suggested that a more favorable response occurs in AD patients who lack filaggrin gene mutations and who exhibit lower total serum IgE levels; however, the optimal dosing of omalizumab for AD has not been determined.9,14 Most recently, Holm and Thomsen stated that “omalizumab is a safe and well-tolerated treatment with some clinical benefit in AD patients. However, the lack of larger [randomized controlled trials] and possible publication bias limit the recommendation of omalizumab for use in clinical practice for AD. Newer and more effective treatments exist and should be prioritized”.17 Before specific recommendations can be made, further research is needed to better define optimal patient selection and dosage recommendations with omalizumab for AD.
Anti-IL-31 Therapy
IL-31 has developed a strong reputation as a mediator of pruritus.8,18,19 Specifically in AD, levels of IL-31 are increased in the skin and blood and correlate with AD severity.8–12,18,19 In both human and murine studies, IL-31 is highly expressed primarily by Th2 lymphocytes, and stimulates its receptors on sensory neurons (C-fibers), dorsal root ganglia, keratinocytes, eosinophils, basophils, and monocytes.3,9–12,18,19 Preliminary data in AD suggests that nemolizumab, a humanized monoclonal antibody against IL-31 receptor A, effectively reduces pruritus, provides some improvements in other clinical manifestations of AD, and appears to exhibit favorable overall safety.3,9–12,19–21
In a Phase II, randomized, controlled, dose-finding, 12-week trial of adults with moderate-to-severe AD inadequately controlled by topical treatments, subcutaneous nemolizumab was compared to placebo.20 In a double-blind, dose-finding, 52-week extension trial (n=131), nemolizumab administered for up to 64 weeks was efficacious and well-tolerated in adults with moderate-to-severe AD inadequately controlled by topical therapy.21 Reductions in the pruritus visual analog scale scores, compared to baseline, were maintained or increased over the duration of study extension (Weeks 12–64), with the greatest therapeutic effect noted in the “0.5mg/kg every four weeks” regimen study arm. The percent change in Eczema Area Severity Index (EASI) score ranged from 68.5 to 78.9 percent depending on the dosage used.21 Further studies are needed with subcutaneous nemolizumab in order to ascertain if this agent will become a rational choice to treat both the visible manifestations and symptomatology associated with AD.
Anti-IL-5 Therapy
IL-5 is a Th2 cytokine that serves as a chemoattractant of eosinophils in inflammation.10 Mepolizumab, a fully humanized monoclonal anti-IL-5 antibody, is FDA-approved for the treatment of severe eosinophilic asthma and eosinophilic granulomatosis with polyangiitis.10,11,22 Thus far, data are limited regarding treatment of AD. Two single, 750-mg doses of subcutaneous mepolizumab, given one week apart, were studied in patients with moderate-to-severe AD using a randomized, placebo-controlled parallel group design (n=40).23 Peripheral blood eosinophil counts were significantly reduced in the mepolizumab-treated group versus in the placebo group (p<0.05). Clinical success was not achieved based on the Physician Global Assessment (PGA) (p=0.115) or Scoring Atopic Dermatitis (SCORAD) measures (p=0.293), changes in pruritus scores, and thymus and activation-regulated chemokine (TARC) serum levels in the mepolizumab-treated group compared to the placebo group.23 At present, due to very limited data, it is not known whether mepolizumab will ultimately offer any therapeutic benefit in AD.
Anti-IL-22 Therapy
Increases in IL-22 have been reported in both acute AD and chronic AD skin lesions.24 Th22 and Th17 cells both produce IL-22, which induces stratum corneum barrier dysfunction and epidermal hyperplasia, exerts a proinflammatory effect in AD lesions, and can promote T-lymphocyte migration into the skin.8
Lesional and nonlesional skin from patients with moderate-to-severe AD (n=59) was evaluated in the context of the administration of fezakinumab, a fully human monoclonal anti-IL-22 antibody, or placebo using transcriptomic and immunohistochemistry analyses.25 A greater reversal of the AD genomic profile was observed in the fezakinumab study group versus placebo (25.3% v. 10.5% at 4 weeks and 65.5% vs. 13.9% at 12 weeks, respectively). Markedly greater mean transcriptomic improvements were seen with fezakinumab in the IL-22-high, fezakinumab-treated group (82.8% and 139.4% at 4 and 12 weeks, respectively) than in the respective IL-22-high, placebo-treated group (39.6% and 56.3% at 4 and 12 weeks) or the IL-22-low groups. Significant downregulations of multiple immune pathways occurred predominantly in patients with high IL-22 levels who received fezakinumab (p<0.05). These data showed that inhibition of IL-22 appears to be most effective in patients with severe AD, exhibiting relevant effects on multiple inflammatory pathways in AD, especially in patients with high IL-22 baseline expression.25
A randomized, double-blind, placebo-controlled trial was performed in patients with moderate-to-severe AD (n=60) treated with intravenous fezakinumab monotherapy or placebo every two weeks for 10 weeks, with follow-up assessments completed through 20 weeks.26 Clinical efficacy was greater in patients with severe AD compared to those with AD of moderate severity. In the patient subset with severe AD, a reduction in SCORAD was significantly greater in the fezakinumab-treated patients than placebo-treated patients at 12 weeks (21.6 vs. 9.6; p=0.029) and 20 weeks (27.4 vs 11.5; p=0.010). Progressive improvements were noted in all efficacy parameters after the last dose (10 weeks) until the end of the study (20 weeks), with upper respiratory tract infections reported as the most frequent adverse event.26 More data are needed, as fezakinumab might be an option for patients with severe AD who exhibit high levels of IL-22 at baseline.
PDE-4 Inhibition
PDE-4 is an intracellular, homeostatic enzyme that regulates proinflammatory or anti-inflammatory effects by modulating the expression of cyclic adenosine monophosphate (cAMP) in several immunologic cell types, including T-lymphocytes, B-lymphocytes, macrophages, eosinophils, monocytes, and neutrophils.3,10,27 The overexpression of PDE-4 has been reported in AD, with mononuclear leukocytes in AD exhibiting increased cAMP-PDE-4 activity that augments inflammation.3,10 The inhibition of PDE-4, via an increase in intracellular cAMP, decreases the transcription and magnitude of several pro-inflammatory cytokines.3,28,29
Apremilast is an oral PDE-4 inhibitor that is currently FDA-approved for the treatment of plaque psoriasis in adults who are candidates for systemic therapy or phototherapy and for active psoriatic arthritis in adults.30 In a pilot study in adult patients with AD (n=16), those receiving apremilast 30mg twice daily showed a significant reduction in EASI score at three months (p=0.008) and six months (p=0.002), Dermatology Life Quality Index (DLQI) at three months (p=0.01) and six months (p=0.03), and visual analog scale scores at six months (p=0.03).31 Gene analyses comparing samples from baseline and during treatment showed modulation in immune response pathways, primarily those related to cAMP-mediated signaling. In a Phase II, double-blind, placebo-controlled, 12-week trial evaluating apremilast 30mg or 40mg twice daily in adults with moderate-to-severe AD (n=185), a statistically significant reduction in EASI score was shown only in the higher-dosage study group; however, this dosage was discontinued by an independent safety monitoring board due to a greater frequency of adverse events.32
Further studies are needed with apremilast and other oral PDE-4 inhibitors in AD.
TSLP-Directed Therapy
Both human and murine studies have shown that TSLP is a key cytokine secreted by epidermal keratinocytes that induces Th2-associated inflammation in atopic disease, including AD, with higher TSLP expression in actively inflamed tissue being an important observation.33 One study of potential AD biomarkers completed in children with AD (n=60) and healthy controls (n=31) revealed that serum levels of TSLP, periostin, and thymus and activation-regulated chemokines (TARC) were higher in children with AD and that TSLP levels might serve as a predictor of more severe AD.34
Tezepelumab is an injectable human monoclonal antibody that blocks the interaction of TSLP with its receptor, thus potentially inhibiting the proinflammatory effects triggered by TSLP in acute and chronic AD lesions.9 The efficacy of tezepelumab in reducing rates of clinically significant exacerbations in patients with poorly controlled asthma has been demonstrated in a Phase II, randomized, double-blind, placebo-controlled trial that evaluated various specified dose levels of tezepelumab versus placebo over a 52-week treatment period.35
A 16-week, Phase IIa study evaluated subcutaneous tezepelumab, compared to placebo, every two weeks in adults with moderate-to-severe AD (n=113), used with a medium-potency topical corticosteroid (TCS).36 With regard to the primary endpoint, a numerically greater percentage of tezepelumab-treated subjects achieved EASI-50 (64.7%) versus placebo-treated subjects (48.2%), although the results were not statistically significant (p=0.091). Treatment-emergent adverse event rates were comparable between the treatment groups. Although not statistically significant, numerical improvements over placebo were noted for all secondary endpoints at Weeks 12 and 16. The greater than expected therapeutic responses observed in the placebo-treated patients might be attributable to use of the TCS.36 Further studies are needed to determine whether tezepelumab has a role in the management of AD.
Concluding Remarks
Cabanillas et al11 accurately noted that “current new therapies under investigation aim to modulate specific inflammatory pathways associated with distinctive atopic dermatitis phenotypes, which would potentially translate into the development of personalized, target-specific treatments of atopic dermatitis.” As the modes of action become focused on specific pathophysiologic pathways, correlations with therapeutic response and potential adverse effects will be further differentiated. Clinicians and their patients will benefit if research is able to develop such target-specific treatments, an effort which is already in progress.
References
- Del Rosso JQ. What’s new in the medicine chest #1. Monoclonal antibody therapies for atopic dermatitis: where are we now in the spectrum of disease management? J Clin Aesthet Dermatol. 2019;12(2):39–43.
- Silverberg JI, Kantor R. The role of interleukins 4 and/or 13 in the pathophysiology and treatment of atopic dermatitis. Dermatol Clinic. 2017;35(3):327–334.
- Rerknimitir P, Otsuka A, Nakashima C, et al. The etiopathogenesis of atopic dermatitis: barrier disruption, immunological derangement, and pruritus. Inflamm Regen. 2017;37:14.
- Czarnowicki T, Krueger JG, Guttman-Yassky E. Novel concepts of prevention and treatment of atopic dermatitis through barrier and immune manipulations with implications for the atopic march. J Allergy Clin Immunol. 2017;139(6): 1723–1734.
- Rudikoff D, Bos JD. Pathophysiology of atopic dermatitis and atopiform dermatitis. In: Rudikoff D, Cohen SR, Scheinfeld N (eds). Atopic Dermatitis and Eczematous Disorders. Boca Raton, FL: CRC Press/Taylor & Francis Group; 2014:107–144.
- Del Rosso JQ, Harper J, Kircik L, et al. Consensus recommendations on adjunctive topical management of atopic dermatitis. J Drugs Dermatol. 2018;17(10):1070–1076.
- Siverberg JI, Silverberg NB. Atopic dermatitis: a heterogenous disorder. Dermatol Clin. 2017;35(3): ix–x.
- Kim JE, Kim JS, Cho DH, et al. Molecular mechanisms of cutaneous inflammatory disorder: atopic dermatitis. Int J Mol Sci. 2016;17(8):1234.
- Deleanu D, Nedelea I. Biologic therapies for atopic dermatitis: an update. Exp Ther Med. 2019;17(2):1061–1067.
- Paller AS, Kabashima K, Bieber T. Therapeutic pipeline for atopic dermatitis: end of the drought? J Allergy Clin Immunol. 2017;140(3):633–643.
- Cabanillas B, Brehler AC, Novak N. Atopic dermatitis phenotypes and the need for personalized medicine. Curr Opin Allergy Clin Immunol. 2017(4);309–315.
- Fabbrocini G, Napolitano M, Megna M, et al. Treatment of atopic dermatitis with biologic drugs. Dermatol Ther. 2018;8(4):527–538.
- Guttman-Yassky E, Dhingra N, Leung DYM. New era of biologic therapeutics in atopic dermatitis. Expert Opin Biol Ther. 2013;13(4):1–23.
- Wang HH, Li YC, Huang YC. Efficacy of omalizumab in patients with atopic dermatitis: a systematic review and meta-analysis. J Allergy Clin Immunol. 2016;138(6):1719–1722.
- Snast I, Reiter O, Hodak E, et al. Are biologics efficacious in atopic dermatitis? a systematic review and meta-analysis. Am J Clin Dermatol. 2018;19(2):145–165.
- Holm JG, Agner T, Sand C, et al. Omalizumab for atopic dermatitis: case series and a systematic review of the literature. Int J Dermatol. 2017;56(1):18–26.
- Holm JG, Thomsen SF. Omalizumab for atopic dermatitis: evidence for and against its use. G Ital Dermatol Venereol. 2019 Feb 4. [Epub ahead of print].
- Mollanazar NK, Smith PK, Yosipovitch G. Mediators of chronic pruritus in atopic dermatitis: getting the itch out?. Clin Rev Allergy Immunol. 2016;51(3):263–292.
- Saleem MD, Oussedik E, D’Amber V, et al. Interleukin-31 pathway and its role in atopic dermatitis: a systematic review. J Dermatolog Treat. 2017;28(7):591–599.
- Ruzicka T, Hanifin JM, Furue M, et al. Anti-interleukin-31 receptor A antibody for atopic dermatitis. N Engl J Med. 2017;376(9):826–835.
- Kabashima K, Furue M, Hanifin JM, et al. Nemolizumab in patients with moderate-to-severe atopic dermatitis: randomized, phase II, long-term extension study. J Allergy Clin Immunol. 2018;142(4):1121–1130.
- Mepolizumab (Nucala) prescribing information. GlaxoSmithKline LLC, Research Triangle Park, NC; 2017.
- Oldhoff JM, Darsow U, Werfel T, et al. Anti-IL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy. 2005;60(5):693–696.
- Ungar B, Garcet S, Gonzalez J, et al. An integrated model of atopic dermatitis biomarkers highlights the systemic nature of the disease. J Invest Dermatol. 2017;137(3):603–613.
- Brunner PM, Pavel AB, Khattri S, et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J Allergy Clin Immunol. 2019;143(1):142–154.
- Guttman-Yassky E, Brunner PM, Neumann AU, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, Phase 2a trial. J Am Acad Dermatol. 2018;78(5):872–881.
- Chan SC, Reifsnyder D, Beavo JA, et al. Immunochemical characterization of the distinct monocyte cyclic-AMP-phosphodiesterase from patients with atopic dermatitis. J Allergy Clin Immunol. 1993; 91(6):1179–1188.
- Souness JE, Aldous D, Sargent C. Immunosuppressive and anti-inflammatory effects of cyclic AMP phosphodiesterase (PDE) type 4 inhibitors. Immunopharmacology. 2000;47(2–3):127–162.
- Grewe SR, Chan SC, Hanifin JM. Elevated leukocyte cyclic-AMP phosphodiesterase in atopic disease: a possible mechanism for cyclic-AMP agonist hyporesponsiveness. J Allergy Clin Immunol. 1982;70(6):452–457.
- Apremilast tablets (Otezla) prescribing information. Celgene Corporation, Summit, NJ; 2018.
- Samrao A, Berry TM, Goreshi R, et al. A pilot study of an oral phosphodiesterase inhibitor (apremilast) for atopic dermatitis in adults. Arch Dermatol. 2012;148(8):890–897.
- Simpson EL, Imafuku S, Poulin Y, et al. A Phase 2 randomized trial of apremilast in patients with atopic dermatitis. J Invest Dermatol. 2018 Dec 5.pii: S0022–202X(18)32905-1. [Epub ahead of print].
- Indra AK. Epidermal TSLP: a trigger for pathogenesis of atopic dermatitis. Expert Rev Proteomics. 2013;10(4):309–311.
- Uysal P, Birtekocak F, Karul AB. The relationship between serum TARC, TSLP and POSTN levels and childhood atopic dermatitis. Clin Lab. 2017;63(7):1071–1077.
- Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377(10):936–946.
- Simpson EL, Parnes JR, She D, et al. Tezepelumab, an antithymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: a randomized Phase 2a clinical trial. J Am Acad Dermatol. 2019;80(4):1013–1021.