
 by Srinivas Sidgiddi, MD, Refika I. Pakunlu, PhD, and Kent Allenby, MD
by Srinivas Sidgiddi, MD, Refika I. Pakunlu, PhD, and Kent Allenby, MD
Drs. Sidgiddi, Pakunlu, and Allenby are with Promius Pharma, a subsidiary of Dr. Reddy’s Laboratories, in Princeton, New Jersey.
Funding: This study was funded and sponsored by the Dr. Reddy’s Laboratories group of companies in Princeton, New Jersy, USA
Disclosures: Drs. Sidgiddi, Pakunlu, and Allenby are employees of Dr. Reddy’s Laboratories, whose subsidiary, Promius Pharma, LLC, markets the topical formulation of betamethasone dipropionate spray, 0.05% (Sernivo™), the product used in this study. Drs. Sidgiddi and Allenby own stock in Dr. Reddy’s Laboratories.
Abstract: Objective: A spray formulation of betamethasone dipropionate 0.05% (BD spray 0.05%; Sernivo™ [betamethasone dipropionate] Spray 0.05%; Promius Pharma, LLC; Princeton, New Jersey, USA) has been developed for the topical treatment of psoriasis. The objective of these studies was to evaluate the efficacy, safety, and potency of BD spray 0.05%. Design, Setting, Participants, and Measurements: Efficacy and safety were assessed in a randomized, vehicle-controlled, double-blind study in adults with moderate plaque psoriasis (ClinicalTrials.gov identifier: NCT01947491). Additionally, the potential for adrenal suppression and systemic absorption was evaluated in a randomized, open-label study in healthy adults (ClinicalTrials.gov identifier: NCT02070965). Potency was measured in two single-point, randomized, evaluator-blinded studies in healthy adults. Results: BD spray 0.05% was significantly more effective than the vehicle spray in subjects with moderate plaque psoriasis after three, 14, and 28 days of twice-daily treatment. The efficacy of BD spray 0.05% was similar to augmented BD lotion 0.05% after 14 days of treatment. The safety of BD spray 0.05% was similar to that of the vehicle spray over 28 days and to that of augmented BD lotion 0.05% over 14 days. Under maximal use conditions for up to 29 days, the potential for adrenal suppression was no greater with BD spray 0.05% than with a 15-day regimen of augmented BD lotion 0.05%. There was less systemic absorption of BD from BD spray 0.05% than from augmented BD lotion 0.05%. Studies classify BD spray 0.05% as a midpotent corticosteroid. Conclusions: BD spray 0.05%, a midpotent corticosteroid, is an effective and well-tolerated treatment for adults with mild to moderate plaque psoriasis.
Keywords: Psoriasis, corticosteroid, betamethasone dipropionate, topical
J Clin Aesthet Dermatol. 2018;11(4):14–22
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease characterized by patches of raised, reddish skin covered by silvery-white scales. It affects about 7.4 million adults (3.2%) in the United States.1 Psoriatic plaques are most commonly found on the scalp, knees, elbows, and lower back, but they can occur anywhere on the body. The plaques are often itchy and painful and can crack and bleed.2 Most patients have limited disease (less than 5% body surface area [BSA] involvement) that can be managed with topical agents,3 including corticosteroids, vitamin D analogues, tar-based preparations, dithranol (anthralin), salicylic acid, and topical retinoids.3,4 Due to the inflammatory nature of the condition, topical steroids are generally recommended as first-line therapy.3
Topical steroids such as betamethasone dipropionate (BD) have been used as an evidence-based treatment for psoriasis and other steroid-responsive dermatoses for more than 40 years.5 Until recently, this synthetic, fluorinated corticosteroid6 was supplied for topical administration in cream, gel, lotion, and ointment formulations. A topical spray formulation of BD 0.05% (Sernivo™ [betamethasone dipropionate] Spray 0.05%; Promius Pharma, a subsidiary of Dr. Reddy’s Laboratories, Princeton, New Jersey, USA) (BD spray 0.05%) is approved for the treatment of mild-to-moderate plaque psoriasis in patients aged 18 years and older.8 BD spray 0.05% uses a vehicle that has high epidermal penetration with reduced absorption into the systemic circulation.7 This article summarizes the results of four studies conducted to evaluate the efficacy, safety, and potency of BD spray 0.05%.
Methods
All studies were conducted according to the United States Code of Federal Regulations Guidelines for Good Clinical Practice [Code of Federal Regulations (21 CFR), Parts 50, 54, 56, 312, and 314]; the International Conference on Harmonisation (ICH) Guidelines for Good Clinical Practice (ICH Guideline E6); the Declaration of Helsinki on the ethical conduct of medical research; and the Belmont Report. Before any procedures were undertaken, subjects were asked to signify their willingness to participate by reading, signing, and dating informed consents. The protocols and informed consent forms were reviewed and approved by the institutional review board operating at each study center.
Efficacy and safety. A Phase III, multicenter, randomized, vehicle-controlled, double-blind, parallel-group study in adults with mild-to-moderate plaque psoriasis was conducted to compare the safety and efficacy of BD spray 0.05% with that of a vehicle and with that of augmented BD lotion 0.05% (Diprolene® AF; Merck & Co., Inc., Kenilworth, New Jersey, USA). (ClinicalTrials.gov identifier: NCT01947491.) The first subject was enrolled on November 8, 2013, and the last subject completed the study on January 12, 2015.
To participate, subjects had to be at least 18 years old; have a clinical diagnosis of stable (3 months or longer) plaque-type psoriasis involving 10 to 20-percent BSA (excluding the face, scalp, groin, axillae, and other intertriginous areas); and have a baseline Investigator’s Global Assessment (IGA) grade of 3 (moderate). Subjects were excluded if they had a current diagnosis of unstable form(s) of psoriasis (including guttate, erythrodermic, exfoliative, or pustular); another inflammatory skin disease that might confound evaluation (e.g., atopic dermatitis, contact dermatitis, tinea corporis); pigmentation; extensive scarring; pigmented lesions; sunburn that might interfere with the rating of efficacy parameters; or a history of psoriasis unresponsive to topical treatments. Also excluded were those with a history of organ transplant requiring immunosuppression, human immunodeficiency virus, or another immunocompromised state. The use of biologic treatments within six months; systemic steroids, phototherapy, or systemic anti-inflammatory agents within 30 days; or topical antipsoriatic drugs, retinoids, or steroids within 14 days of baseline evaluation was prohibited.
Subjects were randomized to receive BD spray 0.05%, vehicle spray, augmented BD lotion 0.05%, or vehicle lotion in a 4:2:2:1 ratio. Subjects in the BD spray 0.05%, vehicle spray, or vehicle lotion groups applied the study drug twice daily for 28 days. To comply with drug labeling restrictions and to maintain blinding, subjects in the augmented BD lotion 0.05% group applied treatment twice daily for 14 days, crossed over to the vehicle lotion group on Day 15, and applied the vehicle lotion twice daily for the subsequent 14 days.
Subject visits occurred at screening to obtain informed consent, tentatively determine eligibility, or to initiate washout; Day 1 (baseline); Day 4?±1 day; Day 8 ±?1 day; Day 15±1 day; and Day 29 ±3 days. The maximum number of days allowed between screening and baseline was 60; the screening and baseline visits could be combined when all required procedures for both visits were completed on the same day. The study duration from initial treatment to final evaluation was 29 days (±?3 days); the maximum duration of subject participation was 92 days (i.e., 60 days for screening plus 32 days after the initial treatment).
Disease severity was assessed primarily by IGA and secondarily by using the Total Sign Score (TSS, the sum of individual scores for erythema, scaling, and plaque elevation) for a representative target lesion on Days 1, 4, 8, 15, and 29. Treatment success was defined as an IGA of 0 or 1 with at least a two-grade reduction from baseline at the Day 15 visit, and the primary efficacy endpoint was the proportion of subjects with treatment success at Day 15. Safety assessments included vital signs (blood pressure, pulse); urine pregnancy tests; local cutaneous safety evaluation of treated areas (atrophy, telangiectasia, itching, pain, and burning/stinging); and adverse events (AEs). Vital signs (blood pressure, pulse) were collected at each visit; urine pregnancy tests for women of childbearing potential were conducted on Days 1, 15, and 29.
All statistical processing was performed using SAS software (SAS Institute, Cary, North Carolina, USA) unless otherwise stated. Two-sided hypothesis testing was conducted for all analyses using a significance level of 0.05. Type-1 error was controlled using a step-wise gatekeeping strategy. The primary efficacy analyses were performed on the intent-to-treat population; safety analyses were performed on the safety population. Detailed descriptions of the methods employed in this study are available elsewhere.8,9
Adrenal suppression. This was a 15-day or 29-day randomized, multicenter, multi-dose, comparator-controlled, open-label study in adults with moderate to severe plaque psoriasis (ClinicalTrials.gov identifier: NCT02070965). Subjects were randomized in a 1:1:1 ratio to receive BD spray 0.05% for 15 days, BD spray 0.05% for 29 days, or augmented BD lotion 0.05% for 15 days. Study treatments were applied twice daily to the affected areas, excluding the face, scalp, groin, axillae, and other intertriginous areas. Subject visits were scheduled at screening; Day 1 (baseline); Days 8, 15, 29 (as appropriate); and Day 43 or 57 (if needed to confirm recovery from hypothalamic-pituitary-adrenal [HPA]-axis suppression).
Clinical determinations of disease severity were conducted using the IGA for overall severity at each visit. No more than 60 days were permitted between screening and baseline. Local cutaneous safety evaluations (including atrophy, telangiectasia, pruritus, pain, and burning/stinging) were performed at each visit. Other safety assessments included vital signs (blood pressure, pulse), urine pregnancy tests, and collection of AEs.
Subjects were tested for HPA-axis function using the adrenocorticotropic hormone (ACTH) stimulation test (intravenous or intramuscular cosyntropin) at least 14 days and no more than 28 days prior to the baseline visit and at the end of treatment visit. The screening ACTH stimulation test was conducted on the day of the screening visit (if timing was appropriate) or on a separate day (e.g., if washout was needed). An abnormal result was defined as a plasma cortisol level of 18?g/dl or less at 30 minutes after ACTH administration. If the HPA axis was suppressed at the end of treatment visit, another test was administered on Day 43 or 57, as appropriate, to confirm recovery, and the test was repeated until the HPA axis was stabilized or explained. At the screening ACTH stimulation test, a blood sample was taken to obtain a baseline value for blood levels (pharmacokinetics [PK]) of BD, betamethasone-17-propionate, and betamethasone. At the end of treatment visit, subjects applied the last dose of the study product at the clinic up to 60 minutes after a pre-treatment PK blood sample was taken; samples were then collected at one, three, and six hours (±5 minutes) after application of the study product to assess the levels of BD, betamethasone-17-propionate, and betamethasone. Plasma BD, betamethasone-17-propionate, and betamethasone were measured by a validated, liquid chromatography-mass spectrometry analytic method with a limit of quantitation of 5pg/mL.
All statistical processing was performed using SAS software version 9.3 or later and all summaries were performed on the safety population. No inferential testing was performed, no interim analyses were planned, and no statistical justification was made for the sample size.
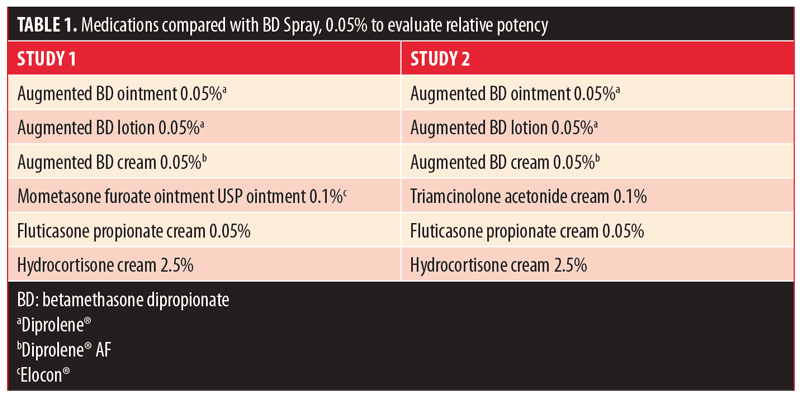
Potency. Two methodologically identical, single-point, randomized, evaluator-blinded, within-subject, single-center studies comparing the vasoconstrictive properties (potencies) of BD spray 0.05% with vehicle spray were conducted (1 pilot and 1 pivotal study). The vasoconstrictive properties were compared with six currently marketed topical steroid formulations of known potency (Table 1) in healthy, non-tobacco-using adults aged 18 to 65 years with a demonstrated blanching response to fluticasone propionate cream 0.05% (Fougera®; E. Fougera & Co., Melville, New York, USA) and a Fitzpatrick skin type of III or less. In both studies, 10?l of each formulation was applied to a single application site on the flexor surfaces of each subject’s forearms (left and right) and kept in place for 16 hours; two untreated control sites were designated on each forearm.
Konica Minolta CR-300 chroma meters (Konica Minolta Sensing Americas, Inc., Ramsey, New Jersey, USA; a-scale reading) were used to measure the degree of vasoconstriction at baseline and at 18 hours postdose using a rating scale in which 0?=?no pallor, 1?=?mild pallor, 2?=?moderate pallor, and 3?=?intense pallor. Assessments were performed under standard fluorescent lighting and at room temperature. The visual evaluators and the chroma meter operators had no knowledge of treatment locations at each site.
Subjects were monitored throughout the studies for AEs, which were collected and subsequently coded in tabular form using the Medical Dictionary for Regulatory Activities version 16.1. The severity of AEs was determined by clinic staff based on the observation and questioning of the affected subjects; investigators judged the relationship of AEs to study treatments.
Statistical analyses were performed separately for the visual and chroma meter data using SAS version 9.3 at a five-percent (two-tailed) significance level. Data were analyzed for mean differences among treatments using a randomized block analysis of variance or a nonparametric analog using the ranks of the scores, as deemed appropriate. The subject was the blocking variable. Within these analyses, pairwise comparisons of the mean assessment scores (chroma meter or visual) were performed using the Ryan–Einot–Gabriel–Welsch Multiple Range Test (REGWQ).
The primary analyses were based on the visual scoring; the secondary analyses used data from the chroma meter corrected for both the average baseline readings and the average baseline-adjusted readings from the untreated sites. The relative potency of the test formulation of BD spray 0.05% was estimated by comparing it with the reference products of known potency.
Results
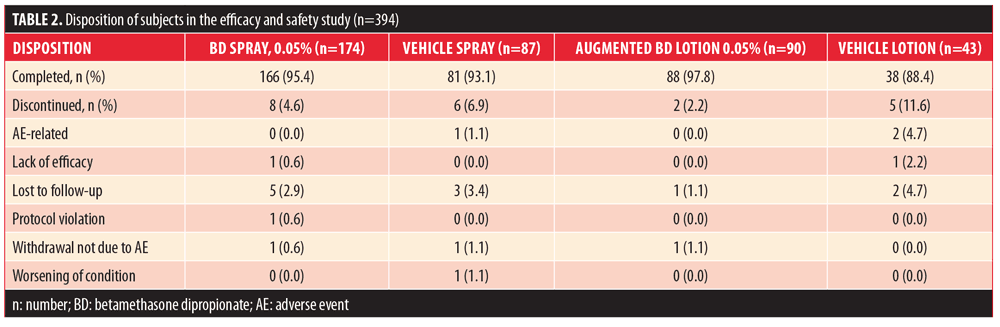
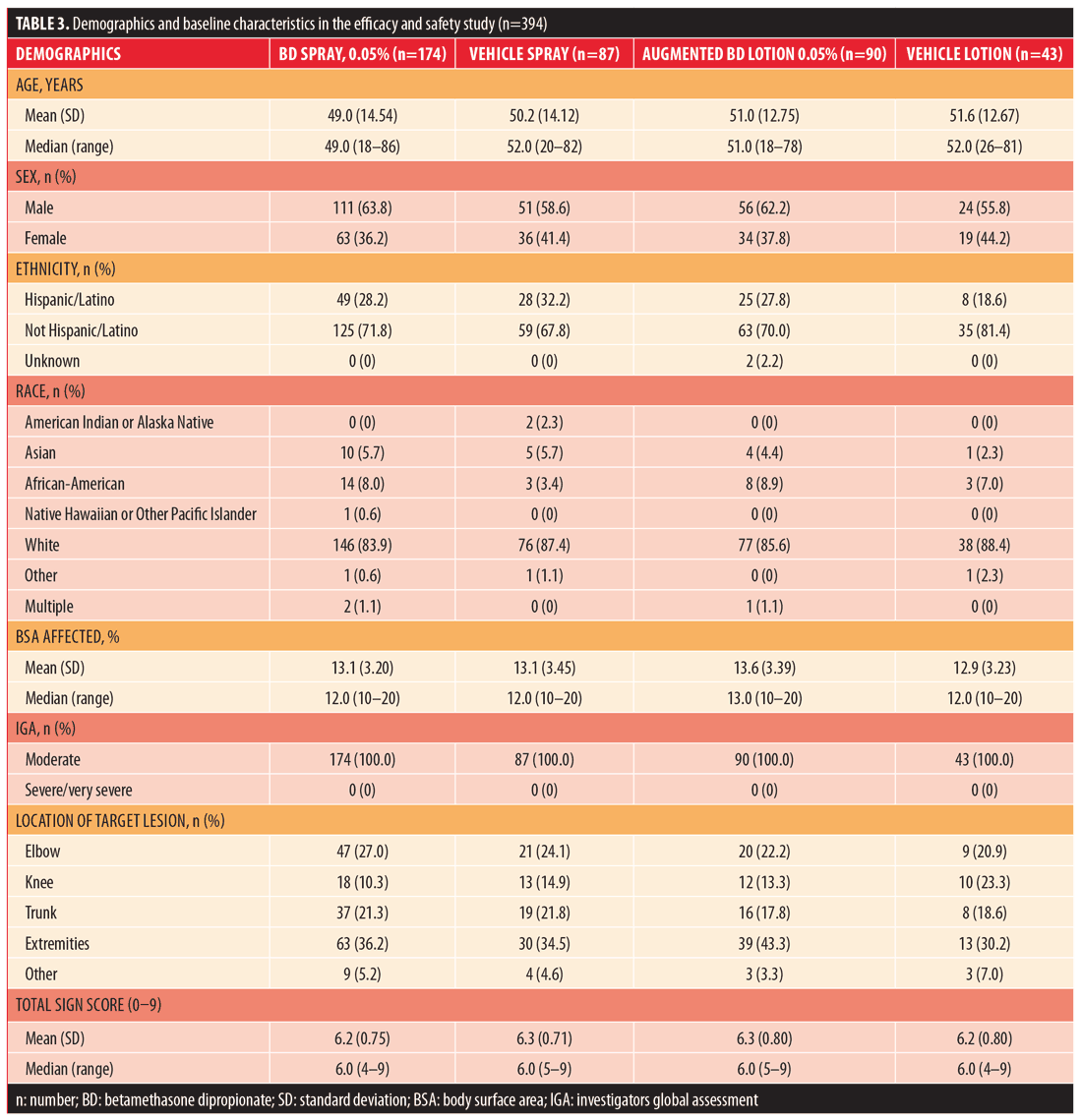
Efficacy and safety. A total of 394 subjects were randomized: 174 to BD spray 0.05%, 87 to vehicle spray, 90 to augmented BD lotion 0.05%, and 43 to vehicle lotion. The majority of subjects (??88%) in all treatment groups completed the study. The most common reason for early withdrawal was subjects being lost to follow-up (Table 2). Demographic and baseline characteristics are presented in Table 3.
For the primary efficacy endpoint, the proportion of subjects with treatment success at Day 15, BD spray 0.05% was significantly superior to vehicle spray (19.0% vs. 2.3%, respectively, P<0.001). The Day 15 treatment success rate for augmented BD lotion 0.05% was 18.9 percent versus 9.3 percent for vehicle lotion. BD spray 0.05% also had significantly greater treatment success than did vehicle spray at Day 8 (10.0% vs. 1.2%, P?= 0.003) and Day 29 (34.5% vs. 13.6%, P?<0.001). Figure 1 shows the rates of success for all study treatments at Days 4, 8, 15, and 29.
BD spray 0.05% significantly outperformed vehicle spray in the percentage of subjects with a reduction of at least 50 percent in the TSS at Day 4 (12.1% vs. 2.3%, P?=?0.004), as shown in Figure 2. At Days 8, 15, and 29, more subjects treated with BD spray 0.05% had reductions of at least 50 percent in the TSS than did subjects in the other treatment groups, but the differences between treatments were not significant.
Safety results over the course of the study are summarized in Table 4. There were no deaths reported in any treatment group. The overall incidence of treatment-emergent AEs (TEAEs) was similar among subjects randomized to BD spray 0.05% and vehicle spray and it was higher among subjects who received augmented BD lotion 0.05% and vehicle lotion.
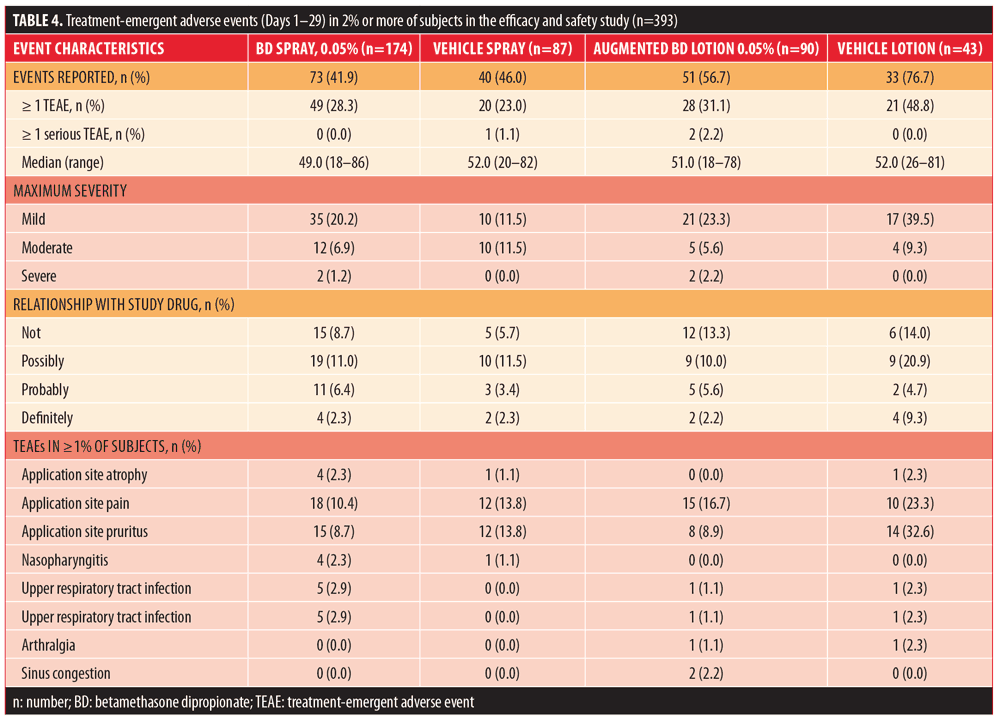
Among subjects treated with BD spray 0.05%, there were no serious TEAEs and no subjects discontinued the study. In the vehicle spray group, one subject (1.1%) reported a serious TEAE (diabetes mellitus, not related to the study treatment) and one subject (1.1%) discontinued the study due to TEAEs of application site pain and application site pruritus (both assessed as probably related to the study treatment). In subjects treated with vehicle lotion, there were no serious TEAEs, but two subjects (4.7%) discontinued the study due to TEAEs of application site pain and application site pruritus (assessed as possibly related to the study treatment). Two serious TEAEs were reported for two subjects (2.2%) in the augmented BD lotion 0.05% group. These serious TEAEs consisted of urinary tract infection, fall, hyponatremia, metabolic acidosis, convulsion, and alcohol withdrawal syndrome in one subject and an exacerbation of chronic obstructive pulmonary disease in the second subject (all assessed as not related to the study treatment). No subjects in the augmented BD lotion 0.05% group discontinued the study.
Among subjects with at least one TEAE in all groups, the majority had a maximum severity of mild. No severe TEAEs were reported in the vehicle spray or vehicle lotion groups. Severe TEAEs (application site pain and application site pruritus) occurred in one subject (0.6%) in the BD spray 0.05% group and in two subjects (1.2%) in the augmented BD lotion 0.05% (one with hyponatremia and alcohol withdrawal syndrome and one with chronic obstructive pulmonary disease).
There were no clinically meaningful changes in vital sign parameters.
Adrenal suppression. In total, 75 subjects were randomized: 23 subjects into 15-day treatment with augmented BD lotion 0.05%, 25 subjects to 15-day treatment with BD spray 0.05%, and 27 subjects to 29-day treatment with BD spray 0.05%. Demographic and baseline characteristics are shown in Table 5.
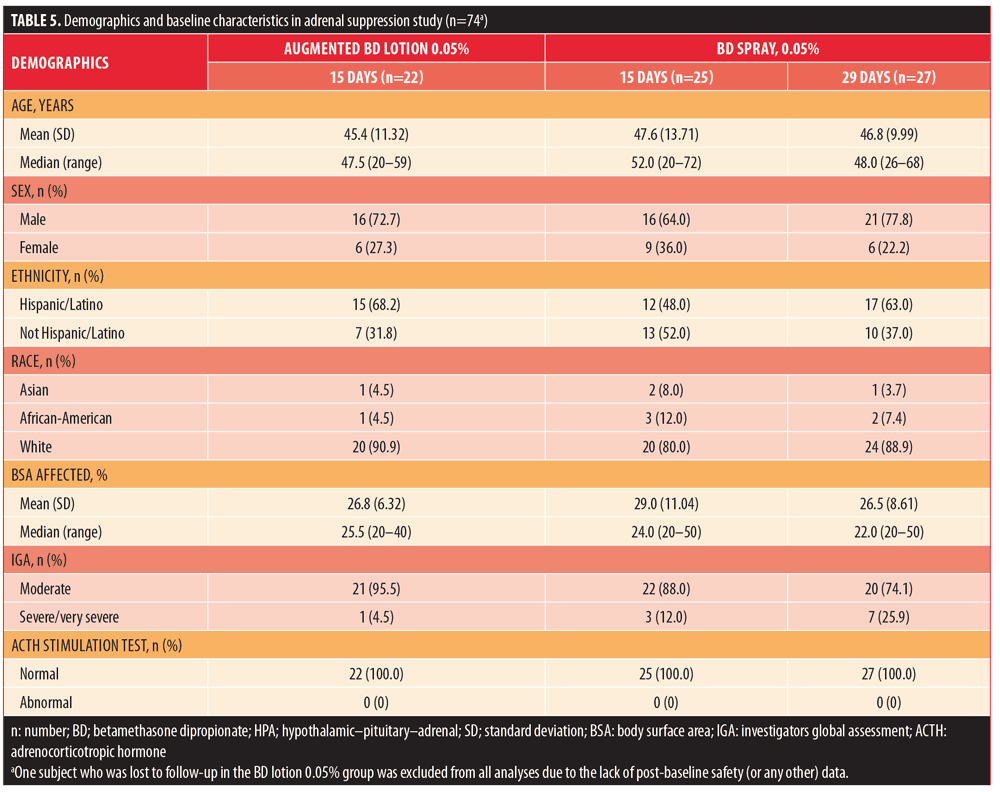
The percentage of subjects who completed the study was similar across the treatment groups (91.3–96.0%). Reasons for discontinuation included subject decision not related to AE (n?= 3, one from each treatment group), lost to follow-up (n?=1, augmented BD lotion 0.05%), and other (n?=1, BD spray 0.05%). All randomized subjects received at least one confirmed dose of study treatment. The subject lost to follow-up in the augmented BD lotion 0.05% group was excluded from all analyses due to the lack of post-baseline data.
A total of 22.7 percent (5/22) of subjects in the BD spray 0.05% 15-day group and 20.0 percent of those in the augmented BD lotion 0.05% 15-day group had abnormal ACTH stimulation test results at the end of treatment that were suggestive of adrenal suppression. No subjects in the BD spray 0.05% 29-day group had abnormal ACTH stimulation test results that were suggestive of adrenal suppression (Table 6).
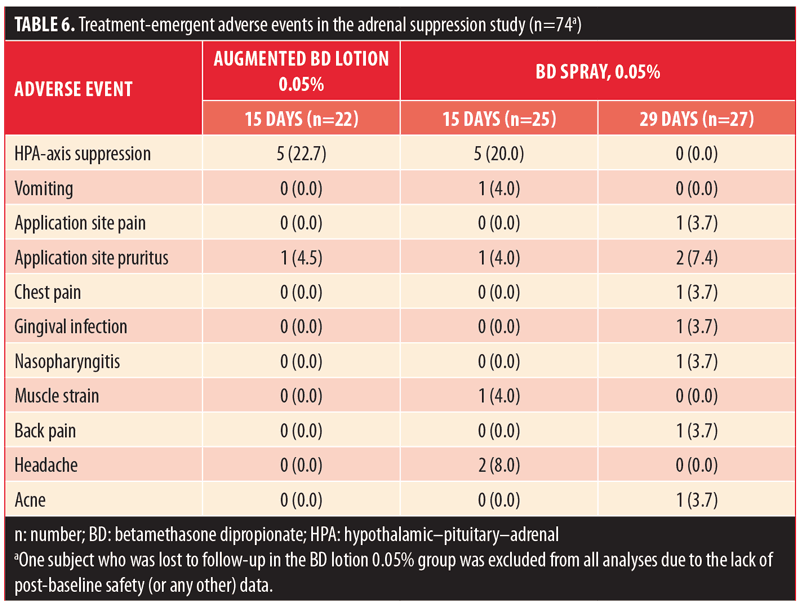
Based on the IGA scores, there was marked improvement in the severity of psoriasis in all three treatment groups (Table 7). By Day 15, the proportion of subjects with almost-clear disease was similar in the BD spray 0.05% and augmented BD lotion 0.05% groups (25% vs. 23.8%, respectively). Notably, 12.5-percent of subjects who were treated with BD spray 0.05% had complete clearance of lesions at Day 15 compared to no subjects treated with augmented BD lotion 0.05%. The overall proportion of subjects with IGA success was 32 percent, 37.5 percent, and 23.8 percent in the BD spray 0.05% (29-day group), BD spray 0.05% (15-day group), and augmented BD lotion 0.05% (15-day group), respectively. (Since treatment with augmented BD lotion 0.05% was not continued beyond Day 14, there were no data to report for the 29-day endpoint.)
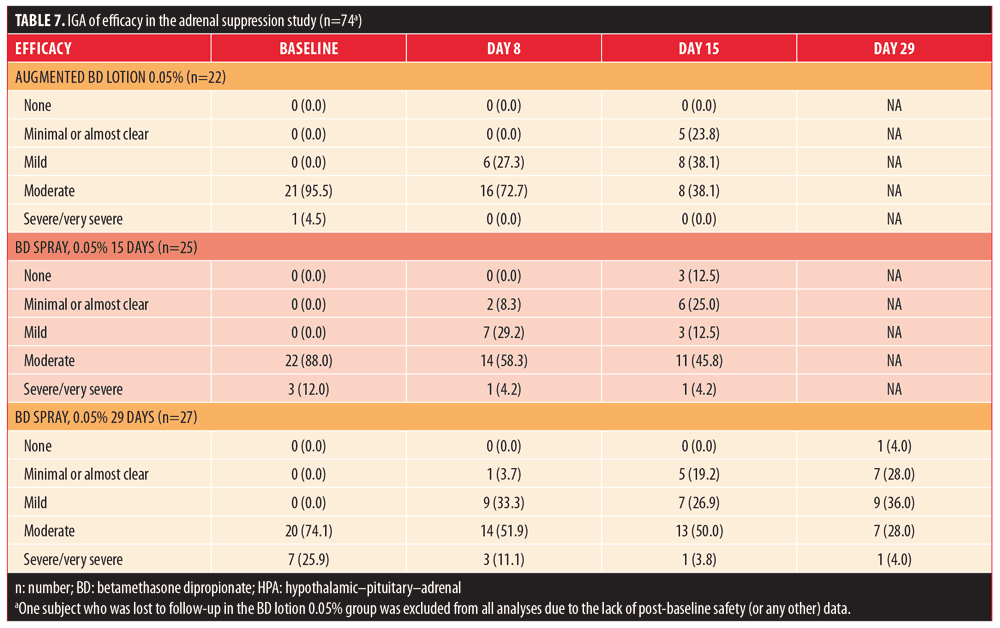
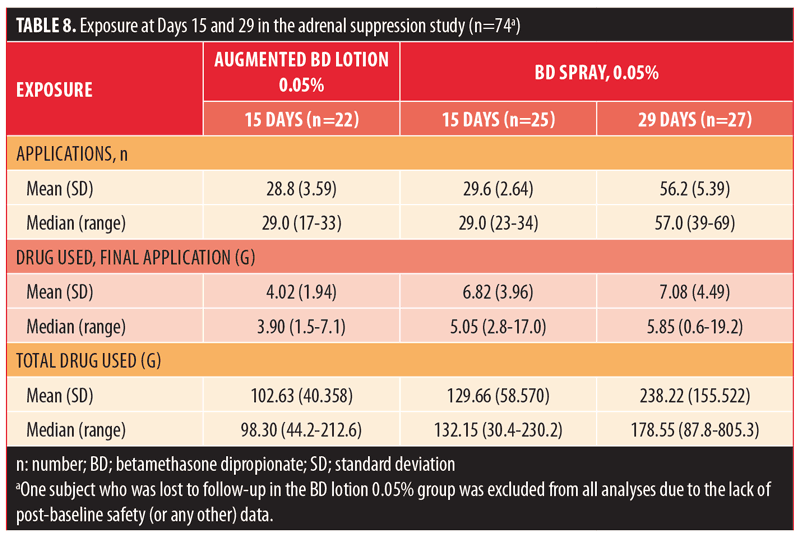
Exposure results are summarized in Table 8. The median number of applications in the 15-day groups was 28 for both BD spray 0.05% and augmented BD lotion 0.05%, while it was 56 in the 29-day group receiving BD spray 0.05%. The maximum number of applications for an individual subject was 43 with augmented BD lotion 0.05% and 67 with BD spray 0.05%. The median amount of study drug used for the final application was greater in the BD spray 0.05% 15-day and 29-day groups (5.05g and 5.85g, respectively) than in the augmented BD lotion 0.05% 15-day group (3.9g). The median total amount of drug used was greater in the BD spray 0.05% 15-day group (132.15g) than in the augmented BD lotion 0.05% 15-day group (98.30g); it was greatest in the BD spray 0.05% 29-day group (178.55g). The maximum amount of study medication used was 212.6g with augmented BD lotion 0.05% and 805.3g with BD spray 0.05%.
The incidence of TEAEs was similar across the treatment groups, ranging from 25.9 to 32 percent. No deaths, serious TEAEs, or discontinuations due to TEAEs were reported. The most frequently reported TEAE was HPA axis suppression in the 15-day treatment groups, followed by application site pruritus in all groups (Table 6). Application site burning/stinging was reported for one subject in the BD spray 0.05% 29-day group. The incidence of burning/stinging, pain, and itching decreased from baseline to day 15 in all groups. Two subjects in the BD spray 0.05% 15-day group reported headaches. No clinically significant atrophy or telangiectasia was seen in any subject at any time during the study and no clinically meaningful changes in vital sign parameters were observed.
The majority of subjects in all groups had no measurable plasma concentration (<?5.00 pg/ml) of betamethasone-17,21-dipropionate at almost all time points just before and after the last application of BD spray 0.05%, whereas the metabolites betamethasone-17-propionate and betamethasone were detected in the majority of subjects. Among subjects with measurable concentrations of betamethasone-17-propionate, the mean concentrations just before the last dose in the augmented BD lotion 0.05% and BD spray 0.05% 15-day groups (100 pg/ml and 117 pg/ml, respectively) were approximately double the concentration seen in the BD spray 0.05% 29-day group (48 pg/ml). A similar trend was seen for the mean of the average of the one-, three-, and six-hour plasma concentrations and the mean Cmax. Results were similar for the other metabolite, betamethasone; the mean concentration at baseline (zero hours) was similar to the mean concentrations at one, three, and six hours after the last dose and similar to the mean Cmax for both metabolites. A comparison of the medians showed that overall metabolite levels were the highest for the augmented BD lotion 0.05% 15-day group.
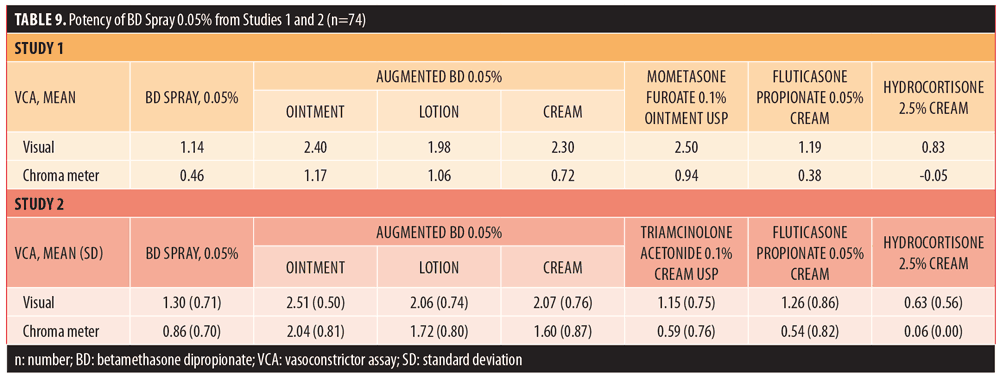
Potency. All 40 subjects enrolled in the pilot potency study completed the study (100%). BD spray 0.05% had mean visual and chroma meter assessment scores of 1.14 and 0.46, respectively, and REGWQ groupings of C,D based on visual assessment and C based on chroma meter data. Using visual data, the potency of BD spray 0.05% fell between that of fluticasone propionate 0.05% cream (midpotent) and hydrocortisone cream USP 2.5% (low-potent). Using the chroma meter findings, the potency of BD spray 0.05% was determined to be between that of augmented BD 0.05% cream (high-potent) and fluticasone propionate 0.05% cream (midpotent). No AEs were reported in this study.
Of 80 subjects enrolled in the pivotal potency study, 78 (97.5%) completed the study. Two subjects (2.5%) did not return to the clinic on Day 2 and were considered to have voluntarily withdrawn. BD spray 0.05% had mean (SD) visual and chroma meter assessment scores of 1.29 (0.71) and 0.86 (0.70), respectively, and a REGWQ grouping of C for both assessments. The visual assessment indicated that the potency of BD spray 0.05% was between that of augmented BD lotion 0.05% (high potent) and fluticasone propionate 0.05% cream (midpotent) and the chroma meter assessment showed that the potency of BD spray 0.05% fell between that of augmented BD cream 0.05% (high potent) and triamcinolone acetonide USP cream 0.1% (upper-midpotent), as shown in Table 9. Two AEs, rash macular and pruritus, were reported by a single subject; both events were considered mild and had resolved.
Discussion
These studies were designed to evaluate the efficacy and safety, potential for adrenal suppression and systemic absorption, and potency of BD spray 0.05%. In the efficacy and safety study, adults with moderate plaque psoriasis treated their condition twice daily for 28 days and BD spray 0.05% was found to be significantly more effective than vehicle spray. The spray formulation also achieved a faster and more pronounced response to treatment than did high-potency augmented BD lotion 0.05% and a vehicle lotion. At the same time, the safety of BD spray 0.05% was similar to that of a vehicle spray over 28 days and to augmented BD lotion 0.05% over 14 days. These findings provide evidence that the novel vehicle technology in BD spray 0.05% allows this midpotent steroid to act more like a high-potent steroid. This suggests that, in clinical practice, BD spray 0.05% might benefit patients with psoriasis as a monotherapy as well as a treatment for resistant plaques in patients receiving systemic therapy.
Results from the adrenal suppression study demonstrate that the potential for HPA-axis suppression with BD spray 0.05% was no greater than with a 14-day regimen of augmented BD lotion 0.05%—even when the spray was used for 28 days. This study also showed that plasma concentrations of BD from BD spray 0.05% were lower than from augmented BD lotion 0.05%, which indicates less systemic absorption. These results were observed despite greater amounts of product used, both in the final application and overall, in the BD spray 0.05% 29-day group than in the augmented BD lotion 0.05% group. Although subjects with abnormal ACTH stimulation test results generally had higher plasma concentrations of betamethasone-17-dipropionate and betamethasone than did subjects with normal results, some subjects with normal post-baseline ACTH stimulation test results had plasma concentrations of betamethasone-17-dipropionate and/or betamethasone as high as some subjects who had abnormal post-baseline ACTH stimulation test results.
The results of the visual assessments and chroma meter measurements in the potency studies indicate that BD spray 0.05% should be classified as a midpotent corticosteroid, based on the Stoughton McKenzie testing methodology10 and REGWQ grouping analyses.
Limitations. Despite many strengths, these studies have some limitations. A larger population in the efficacy and safety study and the HPA-axis suppression study might have allowed for a more meaningful comparison between the groups treated with BD spray 0.05% and augmented BD lotion 0.05%. This limitation, while noteworthy, does not appear to have influenced the results, however.
Conclusion
In subjects with moderate plaque psoriasis, BD spray 0.05% was significantly more effective than vehicle spray, as measured by the proportion of subjects with treatment success (IGA?=?0 or 1 and at least a 2-grade reduction from baseline) after 14 and 28 days of twice-daily treatment, and by a 50-percent or greater reduction in TSS at Days 4, 15, and 29. The spray formulation also yielded an earlier and more pronounced response to treatment than did high-potent augmented BD lotion 0.05% and a vehicle lotion. The safety of BD spray 0.05% was similar to that of a vehicle spray over 28 days and to augmented BD lotion 0.05% over 14 days. With treatment lasting up to 29 days, the potential for adrenal suppression was no greater with BD spray 0.05% than with a 15-day regimen of augmented BD lotion 0.05%. There was less systemic absorption of BD spray 0.05% than of augmented BD lotion 0.05%, as shown by lower median plasma concentrations. Based on visual assessment and chroma meter results from two studies, BD spray 0.05% should be classified as a midpotent corticosteroid.
References
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70(3):512–516.
- National Psoriasis Foundation. About psoriasis. Available at: https://www.psoriasis.org/sites/default/files/psoriasis_fact_sheet.pdf. Accessed July 12, 2017.
- Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis. Section 3. Guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60(4):643–659.
- Mason AR, Mason J, Cork M, et al. Topical treatments for chronic plaque psoriasis. Cochrane Database Syst Rev. 2013;(3):CD005028.
- Nordwall C. Local treatment of psoriasis and eczema with betamethasone-17,21-dipropionate (Diproderm cream): a double-blind comparsion with fluocortolone caproate, fluocortolone pivalate (Ultralanum cream). Curr Ther Res Clin Exp. 1974;16(8):798–803.
- SERNIVO Spray (betamethasone dipropionate) prescribing information. Available at: http://sernivo.com/documents/sernivo-pi.pdf. Accessed July 12, 2017.
- Kircik L, Okumu F, Kandavilli S, Sugarman J. Rational vehicle design ensures targeted cutaneous steroid delivery. J Clin Aesthet Dermatol. 2017;10(2):12–19.
- Stein Gold L, Jackson JM, Knuckles ML, Weiss JS. Improvement in extensive moderate plaque psoriasis with a novel emollient spray formulation of betamethasone dipropionate 0.05. J Drugs Dermatol. 2016;15(3):334–342.
- Fowler JF Jr, Hebert AA, Sugarman J. DFD-01, a novel medium potency betamethasone dipropionate 0.05% emollient spray, demonstrates similar efficacy to augmented betamethasone dipropionate 0.05% lotion for the treatment of moderate plaque psoriasis. J Drugs Dermatol. 2016;15(2):154–162.
- Cornell RC, Stoughton RB. Correlation of the vasoconstriction assay and clinical activity in psoriasis. Arch Dermatol. 1985;121(1):63–67.