
 by Srinivas Sidgiddi, MD; Kent Allenby, MD; Franklin Okumu, PhD; and Anirudh Gautam, MPharm
by Srinivas Sidgiddi, MD; Kent Allenby, MD; Franklin Okumu, PhD; and Anirudh Gautam, MPharm
Drs. Sidgiddi, Allenby, and Okumu are with Promius Pharma in Princeton, New Jersey. Mr. Gautam is with Dr. Reddy’s Laboratories SA in Basel, Switzerland.
FUNDING: This study was funded and sponsored by Dr. Reddy’s Laboratories (DRL #841).
DISCLOSURES: Dr. Sidgiddi, MD is an employee of Dr. Reddy’s Laboratories and owns stock in the company. At the time of the study, Dr. Allenby was an employee of Dr. Reddy’s Laboratories and also owns stock in Dr. Reddy’s Laboratories. At the time of the study, Dr. Okumu was an employee of Dr. Reddy’s Laboratories and owned stock in the company. Mr. Gautam is an employee of Dr. Reddy’s Laboratories and owns stock in the company.
ABSTRACT: Objective. Two clinical studies were conducted to 1) assess the pharmacokinetic (PK) properties of tazarotene and tazarotenic acid in DFD-03 lotion (a 1-minute, short-contact formulation for the topical treatment of acne vulgaris) and tazarotene cream 0.1% and 2) to evaluate transepidermal water loss (TEWL) with DFD-03 lotion, tazarotene gel (0.1%), or vehicle.
Design. The PK study included a single-center, randomized, multiple-dose, laboratory-blinded, open-label, parallel-design, and the TEWL study included a multiple-dose, within-subject comparison design.
Participants. The PK study included healthy adult men aged 18 to 40 years (n=43), and the TEWL study included healthy adults, male or female, aged 18 to 40 years (n=24).
Measurements. PK was assessed via Cmax, AUC0-12, AUC0-24, Tmax, Cmin, Tmin, and fluctuation. TEWL was assessed via evaporimetry.
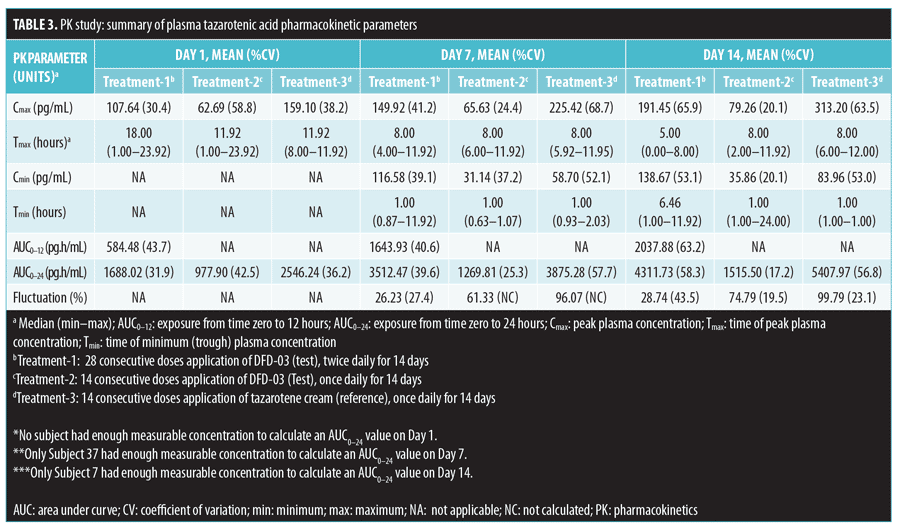
Results. Tazarotene levels were very low due to rapid esterase hydrolysis to the primary active metabolite, tazarotenic acid. Tazarotenic acid AUC0-24 ratios (%) were at least two times higher when the test product was applied twice daily (Treatment-1) versus once daily (Treatment-2) on Days 7 and 14 (268.73% and 254.42%, respectively). Tazarotenic acid AUC0-24 ratios (%) were nearly 100 percent for Treatment-1 versus once-daily tazarotene cream 0.1% (Treatment-3) (99.36% and 83.21%, on Days 7 and 14, respectively). Starting on Day 7, DFD-03 lotion TEWL readings were significantly greater than vehicle (p less than or equal to 0.05), except for on one study day. DFD-03 lotion TEWL readings were numerically greater (nonsignificant) than tazarotene gel.
Conclusion. DFD-03 lotion was well-tolerated, increased TEWL when applied twice daily for one minute, and had a PK profile with similar overall exposure as compared with commercially available tazarotene formulations applied once daily for 12 hours.
KEYWORDS: Pharmacokinetics, acne vulgaris, tazarotene, tazarotenic acid
J Clin Aesthet Dermatol. 2019;12(9):16–24
Acne vulgaris is an inflammatory disease of the skin that occurs in nearly 90 percent of adolescents. Although perceived as a condition experienced exclusively in the teen years, nearly 50 percent of adolescents with acne continue to have symptoms into adulthood, and one percent of men and five percent of women still have acne lesions at age 40 years.1 Acne is associated with negative self-esteem, anxiety, social isolation, and depression. These effects have been shown to be greater in older versus younger adolescents and to increase with age in adults.2–4 Therefore, acne vulgaris is considered a chronic condition, rather than acute, and its treatment can last for decades.1,4,5 Given the prevalence, quality of life burden, and potential need for long-term treatment, there is a substantial patient-driven demand for effective acne therapies, including topical medications that can be used over an extended period.
Retinoids are a class of keratolytic drugs derived from retinoic acid that are used for the treatment of acne and psoriasis. They are comedolytic and anti-inflammatory.1,4,6,7 Topical retinoids are an integral component of acne therapy and are considered appropriate first-line treatments, alone or in combination with antimicrobials, for all but the most severe acne cases.4,7 These drugs are potentially irritating to the skin, with common adverse events (AEs) such as dryness, erythema, stinging, and pruritus, especially over the first few weeks of treatment.6,7 However, formulation advances have led to the removal of potentially irritating surfactants, preservatives, alcohol, and parabens from topical retinoid formulations.8,9
Synthetic retinoids developed for the topical treatment of acne include adapalene, tretinoin, and tazarotene.7 Previous studies have found tazarotene to have equal or greater efficacy compared to tretinoin and adapalene.10–13 Tazarotene is an acetylenic retinoid with well-characterized pharmacology, toxicology, efficacy, and safety.14–18 The primary pharmacologic effects of tazarotene, including normalization of cellular differentiation, decreased hyperkeratinization, reversal of keratinocyte hyperproliferation, and anti-inflammatory effects, contribute to its therapeutic efficacy in treating acne vulgaris. Tazarotene was initially developed as a gel and subsequently in cream and foam formulations.8
Overall, tazarotene has a good safety profile and is not associated with carcinogenicity, contact sensitization, mutagenicity, photoallergic reactions, or phototoxicity.18,19 Topical tazarotene formulations have been associated with local adverse reactions, including desquamation, dry skin, erythema, and burning sensation in approximately 10 to 30 percent of patients, and pruritus, face pain, irritation, and stinging in 1 to 5 percent of patients.16,17,20 Erythema with topical retinoids is due to a physiologic change in the stratum corneum exfoliation process, which leads to a reduction in barrier function, as evidenced by increased transepidermal water-loss (TEWL).18 Often, this increase in TEWL is accompanied by transient accelerated exfoliation (e.g., peeling) and mild irritation.
Tazarotene in gel and cream formulations (Tazorac® Gel and Tazorac® Cream 0.1%, Allergan, Dublin, Ireland) were first approved in the United States in 1997 and 2000, respectively, for the topical treatment of acne vulgaris.21–24 Tazarotene foam (Fabior® Foam 0.1%, Mayne Pharma, Greenville, North Carolina) was approved in 2012 and is indicated for the treatment of acne.25 A pharmacokinetic (PK) study of tazarotene foam 0.1% versus gel 0.1% found that systemic exposure of tazarotene and its active metabolite, tazarotenic acid, in the gel formulation was approximately 1.8 to 2.2 times higher than the foam formulation, based on area under the curve (AUC0–t) and peak plasma concentration (Cmax), yielding lower systemic exposure with the foam.26
DFD-03 lotion, a new short contact formulation containing the active ingredient tazarotene 0.1%, is being developed for use as a twice-daily topical face and body wash for the treatment of acne vulgaris. The formulation was designed to potentially reduce irritation and improve patient convenience, comfort, and treatment adherence. Clinical studies are ongoing to establish the safety and efficacy of DFD-03 lotion in the treatment of acne vulgaris.
This article describes the results of two clinical studies evaluating DFD-03 lotion in normal adult subjects. The first was a PK study comparing the relative bioavailability of tazarotene and tazarotenic acid after application of DFD-03 lotion (once or twice daily) to tazarotene 0.1% cream (once daily). The second study was designed to evaluate the TEWL and appearance of exfoliation and irritation using DFD-03 lotion compared to the commercially available tazarotene 0.1% gel and vehicle lotion.
Methods
Pharmacokinetic study. Objective. The primary objective of this study was to assess the PK parameters of tazarotene and tazarotenic acid following the application of Treatment-1 (DFD-03 lotion twice daily), Treatment-2 (DFD-03 lotion once daily) and Treatment-3 (tazarotene cream 0.1% once daily) and to compare the relative bioavailability of Treatment-1 versus Treatment-2, Treatment-1 versus Treatment-3, and Treatment-2 versus Treatment-3.
Study design and procedure. The study was a single-center, randomized, multiple-dose, laboratory-blinded, open-label, three-arm, parallel-design study conducted in healthy adult male subjects. Two investigational products were administered: the test product, DFD-03 lotion 0.1% (Treatments-1 and -2), and the reference product, tazarotene cream 0.1% (Treatment-3). Treatment-1 was applied twice daily (morning and evening) 12 hours apart. Treatment-2 was applied once daily in the morning. For both Treatments-1 and -2, DFD-03 lotion was rubbed into moistened skin (face, neck, shoulders, upper chest, and upper back) until foamy (approximately 30 seconds), left on for approximately one minute, and then rinsed off with water. For Treatment-3, the reference product was applied to dry skin once daily in the morning, left on for at least 12 hours, and rinsed off. In each study arm, approximately 5g of the test or reference product was applied to the face, neck, shoulders, upper chest, and upper back of the subjects (total area approximately equivalent to 15% body surface area [BSA]) for 14 consecutive days.
Blood samples for the PK measurements were collected on Days 1, 7, and 14. On these days, all subjects were confined to the clinic for at least 10 hours prior to the morning drug application until at least 24 hours after the drug application. The first sample was collected prior to the morning drug application (within 5 minutes before the application for Days 7 and 14), and other samples were collected up to 24 hours after the morning drug application (serial sampling). In the Treatment-1 arm, 11 blood samples were taken on each collection day, for a total of 33 samples in each subject. In the Treatment-2 and Treatment-3 arms, eight blood samples were taken on each collection day, for a total of 24 samples each.
The protocol, its amendments, and the informed consent forms for the PK study were approved by an institutional review board (Advarra, Columbia, Maryland). The study was conducted in compliance with the study protocol and developed based on ethical principles from the Declaration of Helsinki, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use’s Guideline E6 for Good Clinical Practice (GCP), the United States Food and Drug Administration’s GCP Code of Federal Regulations (CFR) Title 21 (part 56), and Directive 2001/20/European Commission. All study participants provided signed informed consent.
Inclusion and exclusion criteria. Subjects were eligible for inclusion in the study if they were 18 to 40 years of age; had a body mass index (BMI) of 18.5kg/m2 to 32.0kg/m2; were light-, non-, or ex-smokers (a light smoker was defined as someone who used nicotine-containing products equivalent to 10 cigarettes or less per day, except e-cigarettes) and had no history of clinically significant diseases or evidence of clinically significant findings on physical examination and/or clinical laboratory evaluations (e.g., hematology, general biochemistry, electrocardiogram, and urinalysis).
Subjects were excluded based on factors that included significant hypersensitivity to tazarotene, retinoids, or any related products (including excipients of the formulations); any skin condition that would interfere with the application of the study medications; use of any dermatological drug therapy within 28 days of first dosing; any significant disease or condition (e.g., gastrointestinal, liver, kidney, cardiovascular, pulmonary, hematologic, neurological, psychiatric, endocrine, immunologic or dermatologic [presence of acne vulgaris was acceptable, but not cystic acne], drug or alcohol abuse, human immunodeficiency virus, hepatitis B, or hepatitis C); use of sun lamps, nonprescription ultraviolet light sources, or tanning beds or excessive exposure to direct sunlight due to pleasure or occupation; maintenance therapy with any drug or significant history of or current drug dependency or alcohol abuse; use of any enzyme-modifying drugs; having taken any investigational product in the 28 days prior to this study; and donation of 500mL or more of blood in the 28 days before Day 1 of this study.
Pharmacokinetic measurements. The PK study samples were assayed for tazarotene and tazarotenic acid at the analytical facility (Algorithme Pharma, Montreal, Canada). The compounds were identified and quantified using high-performance liquid chromatography with mass spectrometry/mass spectrometry detection over a theoretical concentration range of 2.00pg/mL to 500.00pg/mL for tazarotene and 5.00pg/mL to 2,500.00pg/mL for tazarotenic acid. The main PK parameters of interest for this study were Cmax, AUC0–12 (for Treatment-1 only), AUC0–24, and Tmax for Days 1, 7, and 14, and Cmin, Tmin, and fluctuation for Days 7 and 14.
All subjects who provided evaluable data were included in the PK and statistical analysis. Subjects who did not complete the sampling schedule on one of the study days were included in the statistical and PK analyses for only the PK parameters judged to not be affected by the missing sample(s). All reported sampling time deviations following morning drug application on Days 1, 7, and 14 were taken into consideration for the evaluation of PK parameters. When concentrations of tazarotene and tazarotenic acid could not be determined for bioanalytical or clinical reasons, values were set to missing for the statistical and PK analyses.
Absorption and disposition parameters were estimated using a noncompartmental approach. The trapezoidal rule was used to estimate AUC.
Statistical analysis. The primary analysis contained all evaluable subjects. It was estimated that 48 subjects (16 per arm) would be sufficient to meet the study objectives; however, no statistical justification of the sample size was performed.
Descriptive statistics were calculated for plasma concentrations at each time point; for all PK parameters; and to summarize AEs, safety results, and demographic variables (e.g., age, height, weight, and BMI). For all statistical analyses, below the limit of quantitation concentrations occurring prior to the first quantifiable concentration were to be treated as zero and the others set to missing. The natural logarithmic transformation of Cmax and AUC0–24 as well as the rank-transformation of Tmax were used for all statistical inferences, and residuals were calculated as an analysis of variance (ANOVA), adjusting for treatment effects. To evaluate the effect of time on the main PK parameters, Cmax and AUC measures from Day 1, 7, and 14 were included in separate ANOVA models (2-sided analyses; significance set at the 5% level) for each treatment regimen. Dosing time was treated as a continuous variable to estimate the slope on a log-transformed scale. The effect of time on primary PK parameters was assessed based on whether the estimated slope was significantly different from zero. All statistical analyses were conducted using the SAS® version 9 software program (SAS Institute, Cary, North Carolina) using the general linear model procedure for Levene’s test and the mixed procedure for the comparisons of PK parameters between treatments.
Safety. AE data were collected throughout the study. All AEs were classified according to the Medical Dictionary for Regulatory Activities, version 18.0. The assessed safety parameters included the occurrence of AEs, the measurement of clinical laboratory parameters (general biochemistry, hematology, and urinalysis), and vital signs on physical examination. Erythema and scaling were scored for severity as none, mild, moderate, or severe; moderate or severe instances were reported as AEs. Electrocardiograms were recorded when judged necessary by the physician in charge. Clinical laboratory analyses were carried out according to standard procedures of the licensed laboratory, Sanford Health (Sioux Falls, South Dakota).
TEWL study. Objective. The primary objective of this study was to compare the change in facial stratum corneum physiology with the use of DFD-03 lotion 0.1% and tazarotene gel 0.1% versus a vehicle lotion by monitoring TEWL, exfoliation, and irritation over 21 days.
Study design and procedure. This was a randomized, vehicle- and comparator-controlled, assessor-blinded study. Staff who assessed outcomes were blinded to treatment randomization, but staff who applied the study products were not blinded. Treatment assignments were randomized to forehead and cheek sites. Forehead sites were randomized for tazarotene gel and DFD-03 lotion (half of the forehead was randomly assigned to each treatment). On each subject, one cheek was randomly assigned to vehicle lotion and the other cheek served as a no-treatment control. The vehicle lotion was comprised of the same inactive ingredients as DFD-03 lotion without the active ingredient, tazarotene.
Twenty-four subjects were enrolled into this study. DFD-03 and vehicle lotions were applied twice daily to the test sites and removed after one minute with water-dampened cotton balls. The tazarotene gel was applied once daily and removed after five minutes with water-dampened cotton balls. Study product application was repeated for 21 consecutive days.
All test sites were assessed for TEWL using an evaporimeter and visually scored once daily for exfoliation and irritation prior to the morning study product application, at least 60 minutes after any face washing, and at end of study. In addition, test sites were scored for irritation in the evening before study product application. Assessment of exfoliation used the following scale: 0=no visible scaling/peeling; 1=scaling/peeling in a small portion of the test site; 2=up to one-half of the site was peeling or had peeled; and 3=more than one-half of the site was peeling or had peeled. Assessment of irritation/erythema used the following scale: 0=no reaction; 0.5=trace erythema; 1=mild erythema; 2=intense erythema; 3=intense erythema plus edema; and 4=intense erythema plus vesiculation or erosion. If a score of three or more points was observed on either scale, that site was discontinued from further product applications and the last grade for that site was carried forward for all subsequent days. If the study product was discontinued due to severe irritation (but not for exfoliation), subsequent TEWL readings were also discontinued for that test site.
The study protocol, subject recruiting materials, informed consent form, and all other materials provided to subjects were approved by the Bio-Kinetic Clinical Applications Institutional Review Board (Springfield, MO, USA), operating in compliance with FDA GCP CFR Title 21 (part 56). All study participants provided signed informed consent.
Inclusion and exclusion criteria. Individuals were eligible to participate in the study if they met the following criteria: male or female; 18 to 40 years of age at the time of screening; no health conditions (based on pre-study medical history) that would impact their safety during participation; Fitzpatrick skin score of II, III, or IV; if female, agreed to use a study-approved contraceptive method or not be at risk of pregnancy from the screening visit through the end of the study; and, if female, were menstruating on Study Day 1 (or were postmenopausal with no menses for at least one year prior to screening). Additionally, all women participating in the study received pregnancy testing at screening, on Day 1 of treatment, at least once during treatment, and at the end of study.
Subjects were excluded for criteria that included the use of any investigational product within 30 days prior to the study; sensitivity/allergy to the study product ingredients or known history of adverse reactions to any topical formulation; diabetes mellitus, asthma, hypertension or circulatory disease; use of any corticosteroids (by any route) or oral retinoids within 14 days prior to screening visit; change in (including initiation/ending) dose of any prescription or over-the-counter medication; use of prescription or over-the-counter topical medications on the planned test sites; clinically significant allergy to soaps, lotions, emollients, ointments, creams, cosmetics, adhesives, or latex; significant skin conditions or disorders (e.g., psoriasis, atopic dermatitis, porphyria, vitiligo), skin anomalies such as a recent sunburn, scratch, scar tissue, tattoo, or coloration; history of dermatologic cancers; if female, if pregnant, lactating, breastfeeding, or intended to become pregnant; and drug or alcohol addiction or abuse in the past one year.
Statistical analysis. All available data were included in the analysis of the pharmacodynamic variables. No imputation was conducted for missing data except when the last observation was carried forward when the study product was discontinued due to severe irritation.
TEWL readings were tabulated, including an arithmetic mean, standard deviation, median, and range (minimum, maximum) for each study day. Treatments were compared to each other for each study day using Student’s t test, and maximum TEWL readings were compared. These analyses were performed separately for each facial site.
Daily morning scores for irritation and exfoliation were summed for a total score for each subject, and study products were compared using Student’s t test and one-way ANOVA followed by Dunnett’s test. The day to reach maximum exfoliation (score of 3 points) was also compared. If a score of three points was never achieved, then the day was set to 22. These analyses were performed separately for each facial site.
Safety. Participants were monitored throughout the duration of the study for AEs. Each sign or symptom was reviewed and assessed for severity and any relationship to the study product, with the date and time of onset, duration, and resolution recorded. Signs of exfoliation or irritation were not recorded as AEs unless they led to early study product discontinuation.
Test sites were scored once daily for exfoliation and twice daily for irritation. All subjects who received at least one application of the study product on Day 1 were included in the safety population.
Results
Pharmacokinetic study. Subject disposition and demographics. A total of 43 subjects were included; after randomization, 14 subjects received Treatment-1, 14 received Treatment-2, and 15 received Treatment-3. Two subjects (5%) discontinued the study: one withdrew for personal reasons (not related to clinical events) and one was withdrawn due to a positive drug test result. A total of 41 subjects (95%) completed the study. All 43 subjects were analyzed and included in the PK and statistical analysis, and the safety population included all subjects who entered the study and received at least one investigational product. Subject demographic characteristics are summarized in Table 1.
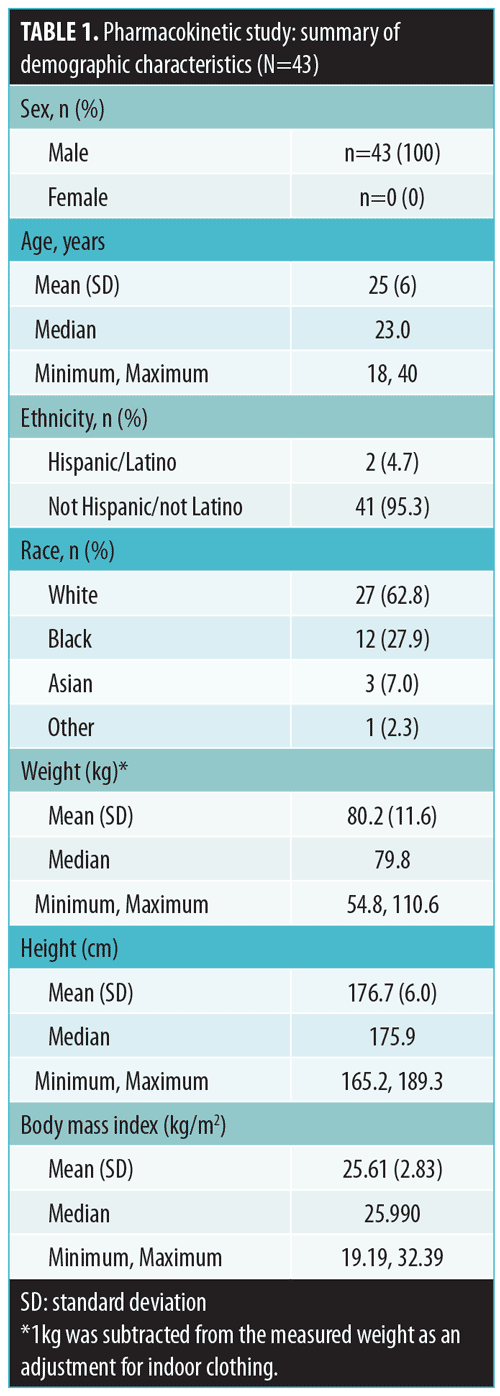
Tazarotene. Mean plasma concentration-time profiles on Days 1, 7, and 14 are displayed by treatment in Figure 1. Plasma levels were below the lower limit of quantification (LOQ: 2.00pg/mL) in all samples collected prior to first dosing on Day 1.
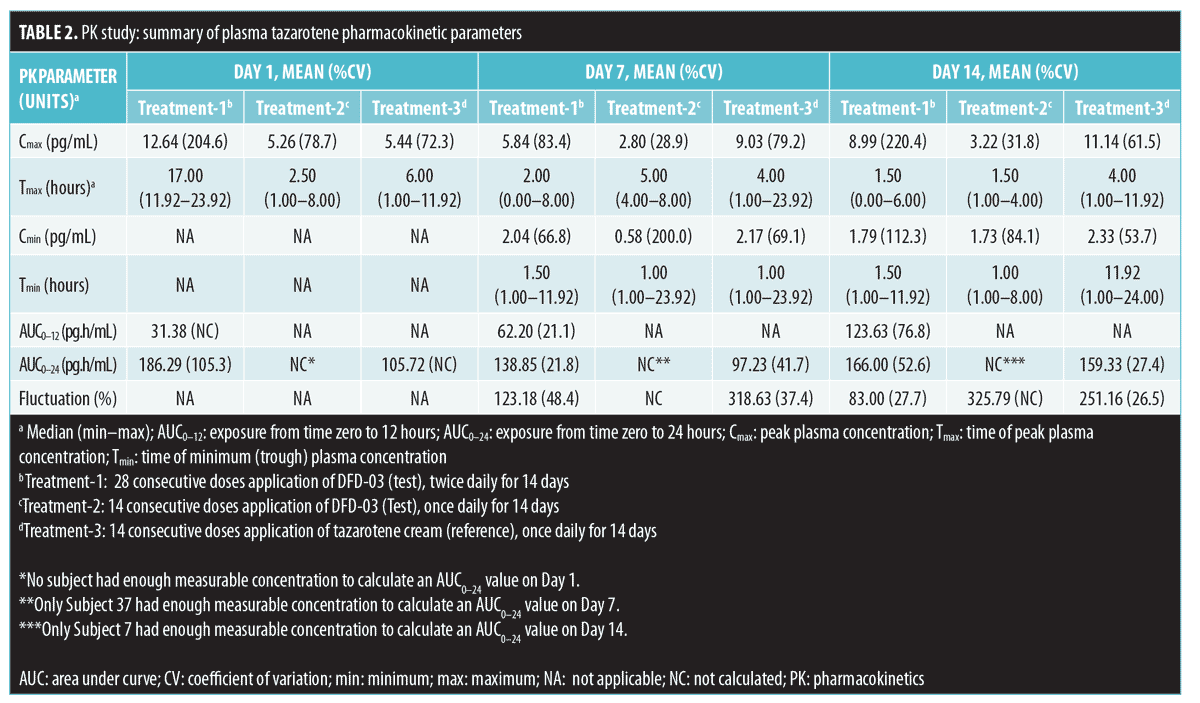
As expected, very low concentrations of the parent compound, tazarotene, were detected in plasma samples. Following topical application, tazarotene undergoes rapid esterase hydrolysis to its primary active metabolite, tazarotenic acid, and typically little parent compound is detected in plasma.14 On Day 1, following twice-daily dosing of the test product (Treatment-1), 10 subjects had at least one measurable concentration above the LOQ as compared with four subjects for the once-daily dosing regimen (Treatment-2) and 13 subjects receiving once-daily dosing with the reference product (Treatment-3). On Day 7, the numbers of subjects with at least one measurable concentration above the LOQ were 13, four, and 14 for Treatment-1, Treatment-2, and Treatment-3, respectively. On Day 14, the numbers of subjects with at least one measurable concentration above the LOQ were 14, six, and 13 for Treatment-1, Treatment-2, and Treatment-3, respectively. Tazarotene PK parameter values by treatment arm for Days 1, 7, and 14 are presented in Table 2. Mean Cmax values ranged from 3 to 13pg/mL. Most of the concentration values were below the LOQ of 2.00pg/mL; as a result, PK parameters for this analysis are to be interpreted with caution.
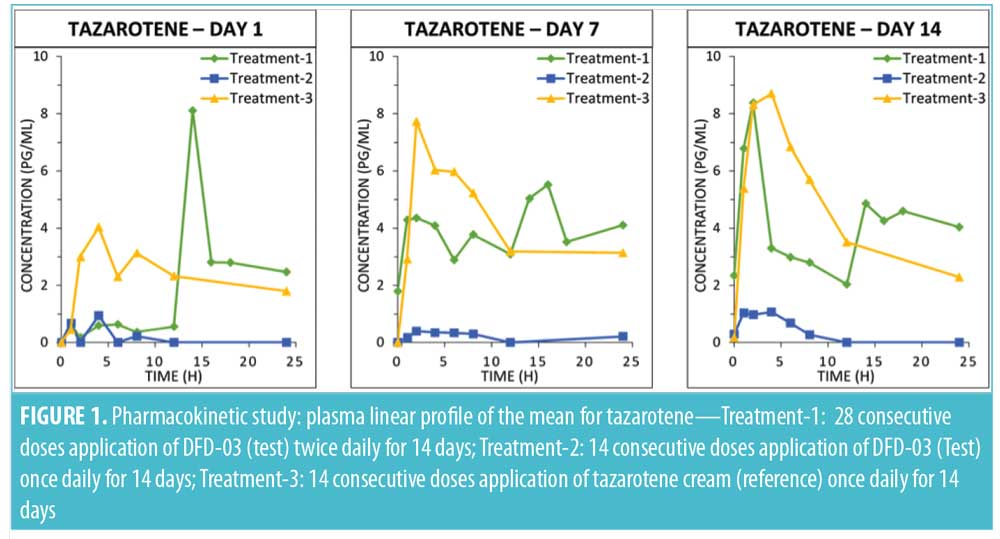
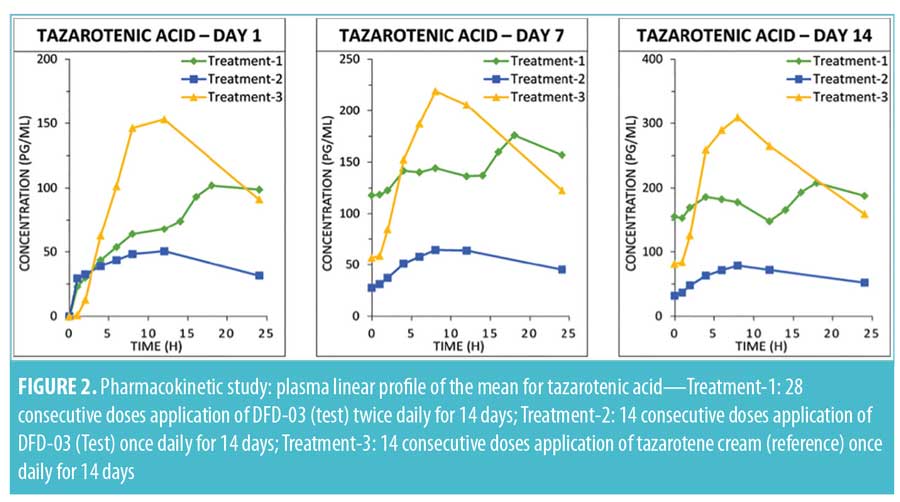
Tazarotenic acid. Mean plasma concentration-time profiles on Days 1, 7, and 14 are displayed by treatment in Figure 2. As with tazarotene, plasma levels for tazarotenic acid were below the LOQ (5.00 pg/mL) in all samples collected prior to the Day 1 first dosing.
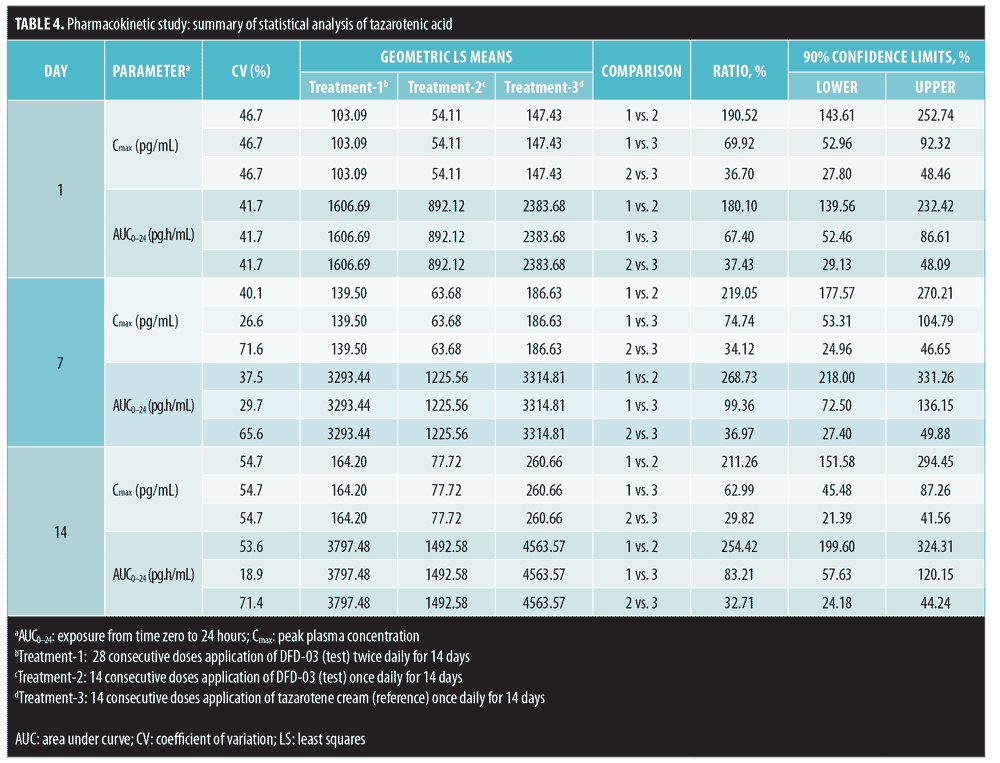
A summary of PK parameter values for tazarotenic acid, by treatment for Days 1, 7, and 14, is presented in Table 3, and a summary of the statistical analyses of Cmax and AUC is given in Table 4. Reflecting the different dosing regimens, exposure to tazarotenic acid was about two times higher when the test product was applied twice daily (Treatment-1), compared to once daily (Treatment-2), with Cmax ratios of 190.52 percent, 219.05 percent, and 211.26 percent, and AUC0–24 ratios of 180.10 percent, 268.73 percent, and 254.42 percent for Days 1, 7, and 14, respectively. The data also largely show that exposure to tazarotenic acid was less when the product was applied as a lotion once daily (Treatment-2) or twice daily (Treatment-1) than as a cream applied once daily (Treatment-3). Exceptions to this observation occurred on Days 7 and 14, where the ratios of least squares means for Treatment-1 versus 3 AUC0–24 values were close to 100 percent, with values of 99.36 percent and 83.21 percent, respectively. The slope estimates for the PK parameters as a function of time (Days 1, 7, and 14) were close to zero for all PK parameters and treatments.
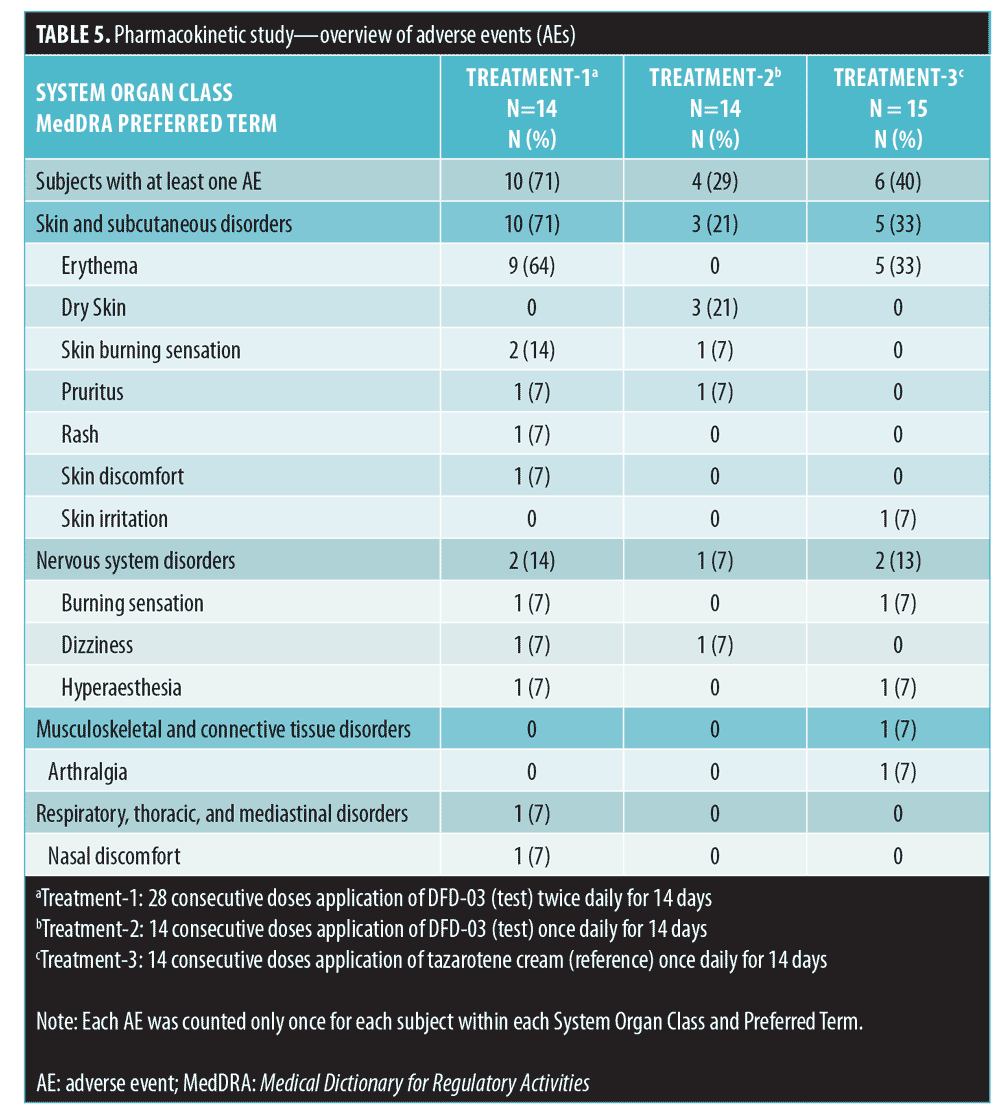
Adverse events. Of the 43 subjects analyzed, 20 (47%) reported a total of 41 AEs over the course of the study (Table 5). All AEs reported during the study were mild in severity; no moderate or severe AEs were reported. Subjects who received twice-daily treatment with the test product (Treatment 1) reported AEs with a higher incidence, compared to those receiving once-daily application of either the test or reference product (Treatment-2 and Treatment-3). Incidence rates of AEs in the three treatment groups were 71 percent, 29 percent, and 40 percent, respectively. Similarly, the Treatment-1 group had the highest incidence of drug-related AEs, with 10 of 14 subjects (71%) reporting 24 AEs, the most common of which was erythema (9 subjects; 64%). The next highest incidence was in the Treatment-3 group, with six of the 15 subjects (40%) reporting 11 AEs, the most common of which was erythema (5 subjects; 33%). In the Treatment-2 group, four of 14 subjects (29%) reported six AEs, the most common of which was dry skin (3 subjects; 21%). No serious AEs or deaths were reported for any subjects enrolled in this study, and no subject was withdrawn for safety reasons.
TEWL study. Subject Disposition and Demographics. A total of 24 subjects were enrolled, five women and 19 men. The mean age was 29.5 years, with a range of 22 to 40 years. All subjects were white; one subject was Hispanic. Subject demographic characteristics can be found in Table 6.

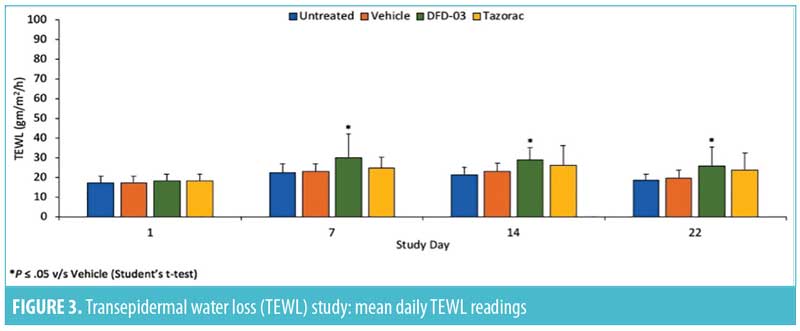
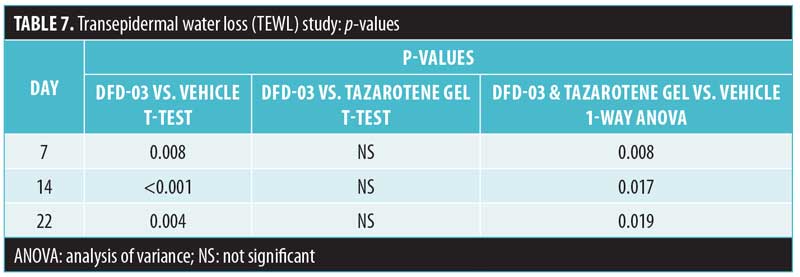
Transepidermal water loss. Mean daily TEWL readings are shown in Figure 3, and TEWL P-values are shown in Table 7. Starting at Day 7, TEWL readings for sites treated with DFD-03 lotion were significantly greater than those treated with vehicle lotion (p less than or equal to 0.05), except for Day 21, where the difference was numerically greater. The effect on TEWL readings with DFD-03 lotion were numerically greater than the tazarotene gel-treated sites; however, these differences were not statistically significant, except for Day 17 where p=0.048.
Safety. There were four AEs reported in four (16.7%) subjects. Two subjects reported a mild stinging sensation lasting one day, one subject reported a mild burning sensation lasting two days, and one subject had broken capillaries in the right eye lasting six days.
Across all products, mean daily irritation scores ranged from 0 to 0.5 points. When daily scores were summed for each patient, the mean total irritation score for the untreated cheek site was 5.5 points. The mean total irritation score for DFD-03 lotion was 1.8 points, compared to 5.9 points for the vehicle lotion (p<0.001). The mean total irritation score for tazarotene gel was 1.1 points (p<0.001 for tazarotene gel vs. vehicle lotion). One-way ANOVA was p<0.001 for DFD-03 and tazarotene gel versus vehicle lotion.
Across all products, mean daily exfoliation scores ranged from 0 to 0.2 points. There was no statistically significant difference between the summed mean total exfoliation scores for either DFD-03 lotion or tazarotene gel as compared with vehicle lotion (p = 0.445 and p = 0.624, respectively). Mean total exfoliation scores were 2.2, 1.7, 1.0, and 0.2 points for DFD-03 lotion, tazarotene gel, vehicle lotion, and untreated, respectively.
Discussion
Together, these two studies of DFD-03 (tazarotene) lotion (0.1%) compared to tazarotene cream or gel (0.1%) for the topical treatment of acne vulgaris suggest that this new formulation has an expected PK profile with similar overall exposures for twice-daily DFD-03 and once-daily tazarotene. Twice-daily application of DFD-03 was well-tolerated and showed similar retinoid activity as assessed by TEWL.
Tazarotene is known to undergo rapid esterase hydrolysis to its primary active metabolite, tazarotenic acid. In the PK study, the DFD-03 lotion behaved as expected, leaving little detectable parent compound in the plasma. Mean Cmax values for tazarotene were largely below the limit of quantitation; therefore, the PK parameters must be interpreted with some caution. However, the PK of DFD-03 lotion appeared to be linear. In accordance with the dosing regimen, the exposure to tazarotenic acid was about two times higher when the test product was applied as a lotion twice daily (Treatment-1), compared to once daily (Treatment-2). In addition, exposure to tazarotenic acid was similar overall when the test product, DFD-03 lotion, was applied twice daily (Treatment-1), compared to the reference product (tazarotene cream), which was applied once daily (Treatment-3). In the TEWL study, DFD-03 lotion-treated sites had significantly increased TEWL readings compared to vehicle lotion-treated sites, suggesting that DFD-03 lotion has significant retinoid activity. DFD-03 lotion was also associated with numerically greater TEWL readings than tazarotene gel, but this difference was not statistically significant.
Overall, DFD-03 lotion was well-tolerated in the PK study. AEs occurred in less than one-half (47%) of subjects, and all reported AEs were mild in severity. Subjects in the Treatment-1 arm reported AEs with a higher incidence than the Treatment-2 and Treatment-3 arms (71%, 29%, and 40%, respectively); the majority of these were treatment-related (71%, 21%, and 40%, respectively). The most common AE, erythema, was observed in 64 percent of patients using the DFD-03 lotion twice daily (Treatment-1) and 33 percent of patients using the reference treatment (Treatment-3). However, there were no reported instances of erythema in patients using the test product once daily (Treatment-2). Additionally, the highest severity reported for erythema was moderate. In the TEWL study, AEs were reported in four (16.7%) subjects; all were mild and resolved over a few days. The mean irritation score was significantly lower for subjects using the DFD-03 lotion, compared to those using the vehicle lotion, and mean exfoliation score was similar across all treatment groups, including the vehicle group. The lower irritation scores for DFD-03, compared to the vehicle, are probably due to the natural rosy coloration of the cheeks (vehicle site), compared to the forehead (DFD-03 site), rather than any treatment effect. Overall irritation and exfoliation levels were less than expected, with most subjects experiencing no irritation or exfoliation with either retinoid-containing product. There were no deaths or serious AEs in either the PK or the TEWL studies, and no subject was withdrawn from either study due to safety reasons.
DFD-03 (tazarotene) lotion performed similarly to tazarotene cream (in the PK study) and tazarotene gel (in the TEWL study). In the PK study, overall exposure based on Cmax and AUC0–24 values for tazarotene and tazarotenic acid for DFD-03 lotion administered once-daily (Treatment-2) were lower than tazarotene cream once-daily on Days 1, 7, and 14, and the values for DFD-03 lotion administered twice daily were similar or somewhat lower than tazarotene cream. The AE rate was higher in the DFD-03 lotion twice-daily group than in the tazarotene cream once-daily group, but the DFD-03 lotion once-daily group had a lower incidence of AEs than both the other groups. In the TEWL study, the TEWL readings for DFD-03 lotion were greater than for tazarotene gel, although the difference was not statistically significant.
Limitations. Due to the documented PK profile of tazarotene, DFD-03 lotion was expected to leave very low concentrations of the parent compound in plasma samples, and results were consistent with this expectation. However, because most of the concentration values were below the limit of quantification, PK parameters for tazarotene are to be interpreted with caution. In addition, including only male subjects in the PK study limits the applicability of the results to the female population, although plasma tazarotenic acid concentrations are known to be independent of sex.23,24
Conclusion
Multiple tazarotene formulations have shown efficacy in treating acne vulgaris. These include tazarotene plus other active ingredients (e.g., clindamycin, benzoyl peroxide, dapsone)27–29 and tazarotene foam, cream, and gel.8,15–17,26,30–32 Recently, novel formulations have been developed that aim at improving tazarotene topical delivery while reducing side effects.9,33 DFD-03 lotion is a new, short-contact tazarotene formulation designed to increase comfort and convenience, potentially reduce irritation, and improve patient adherence and outcomes. The results of these studies showed that DFD-03 lotion was well-tolerated and, based on increased TEWL with twice-daily use, suggest its effectiveness. Additionally, DFD-03 lotion appears to have a PK profile with similar overall exposure, as seen in existing tazarotene formulations.
Acknowledgments
The authors would like to thank Amber Anderson, Certified Clinical Research Coordinator, and Brendon Bourg, Vice president and General Manager, from QPS Bio-Kinetic for assisting with the study conduct. The authors would also like to thank Paul Lehman, MSc, Vice President and Head of Transdermal Research Services, from QPS LLC for his scientific expertise in the TEWL study.
References
- Dawson AL, Dellavalle RP. Acne vulgaris. BMJ. 2013;346:f2634.
- Lasek RJ, Chren MM. Acne vulgaris and the quality of life of adult dermatology patients. Arch Dermatol. 1998;134(4):454–458.
- Tasoula E, Gregoriou S, Chalikias J, et al. The impact of acne vulgaris on quality of life and psychic health in young adolescents in Greece. results of a population survey. An Bras Dermatol. 2012;87(6):862–869.
- Thiboutot D, Gollnick H, Bettoli V, et al. New insights into the management of acne: an update from the Global Alliance to Improve Outcomes in Acne group.
J Am Acad Dermatol. 2009;60(5 Suppl):S1–S50. - Gollnick HP, Finlay AY, Shear N, Global Alliance to Improve Outcomes in Acne. Can we define acne as a chronic disease? If so, how and when? Am J Clin Dermatol. 2008;9(5):279–284.
- Thielitz A, Gollnick H. Topical retinoids in acne vulgaris: update on efficacy and safety. Am J Clin Dermatol. 2008;9(6):369–381.
- Zaenglein AL, Pathy AL, Schlosser BJ, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2016;74(5):945–973.e33.
- Gregoriou S, Kritsotaki E, Katoulis A, Rigopoulos D. Use of tazarotene foam for the treatment of acne vulgaris. Clin Cosmet Investig Dermatol. 2014;7:165–170.
- Aggarwal G, Nagpal M, Kaur G. Development and comparison of nanosponge and niosome based gel for the topical delivery of tazarotene. Pharm Nanotechnol. 2016;4(3):213–228.
- Leyden JJ, Tanghetti EA, Miller B, et al. Once-daily tazarotene 0.1 % gel versus once-daily tretinoin 0.1 % microsponge gel for the treatment of facial acne vulgaris: a double-blind randomized trial. Cutis. 2002;69(2 Suppl):12–19.
- Pariser D, Colon LE, Johnson LA, Gottschalk RW. Adapalene 0.1% gel compared to tazarotene 0.1% cream in the treatment of acne vulgaris. J Drugs Dermatol. 2008;7(6 Suppl):s18–23.
- Thiboutot D, Arsonnaud S, Soto P. Efficacy and tolerability of adapalene 0.3% gel compared to tazarotene 0.1% gel in the treatment of acne vulgaris. J Drugs Dermatol. 2008;7(6 Suppl):S3–S10.
- Webster GF, Berson D, Stein LF, et al. Efficacy and tolerability of once-daily tazarotene 0.1% gel versus once-daily tretinoin 0.025% gel in the treatment of facial acne vulgaris: a randomized trial. Cutis. 2001;67(6 Suppl):4–9.
- Tang-Liu DD, Matsumoto RM, Usansky JI. Clinical pharmacokinetics and drug metabolism of tazarotene: a novel topical treatment for acne and psoriasis. Clin Pharmacokinet. 1999;37(4):273–287.
- Shalita AR, Chalker DK, Griffith RF, et al. Tazarotene gel is safe and effective in the treatment of acne vulgaris: a multicenter, double-blind, vehicle-controlled study. Cutis. 1999;63(6):349–354.
- Shalita AR, Berson DS, Thiboutot DM, et al. Effects of tazarotene 0.1 % cream in the treatment of facial acne vulgaris: pooled results from two multicenter, double-blind, randomized, vehicle-controlled, parallel-group trials. Clin Ther. 2004;26(11):
1865–1873. - Feldman SR, Werner CP, Alio Saenz AB. The efficacy and tolerability of tazarotene foam, 0.1%, in the treatment of acne vulgaris in 2 multicenter, randomized, vehicle-controlled, double-blind studies. J Drugs Dermatol. 2013;12(4):438–446.
- Chandraratna RA. Tazarotene—first of a new generation of receptor-selective retinoids. Br J Dermatol. 1996;135 Suppl 49:18–25.
- Hogan DJ, Saenz AB. Phototoxic and photoallergic potential of tazarotene foam 0.1% in 2 phase 1 patch studies. Cutis. 2012;90(5):266–271.
- Kang S, Leyden JJ, Lowe NJ, et al. Tazarotene cream for the treatment of facial photodamage: a multicenter, investigator-masked, randomized, vehicle-controlled, parallel comparison of 0.01%, 0.025%, 0.05%, and 0.1% tazarotene creams with 0.05% tretinoin emollient cream applied once daily for 24 weeks. Arch Dermatol. 2001;137(12):1597–1604.
- Drug approval package. Tazorac (tazarotene) cream. United States Food & Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21-184_Tazorac.cfm. Accessed 23 Apr 2018.
- Drug approval package. Tazorac (tazarotene) gel. United States Food & Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/nda/97/020600_tazorac_toc.cfm. Accessed 23 Apr 2018.
- Tazorac (tazarotene) cream, 0.05%/0.1% [prescribing information]. Irvine, CA: Allergan; 2017.
- Tazorac (tazarotene) gel, 0.05%/0.1% [prescribing information]. Madison, NJ: Allergan; 2018.
- Drug approval package. Fabior (tazarotene) foam, 0.1%. United States Food & Drug Adminstration website. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202428_fabior_toc.cfm. Accessed 23 Apr 2018.
- Jarratt M, Werner CP, Alio Saenz AB. Tazarotene foam versus tazarotene gel: a randomized relative bioavailability study in acne vulgaris. Clin Drug Investig. 2013;33(4):283–289.
- Maiti R, Sirka CS, Ashique Rahman MA, et al. Efficacy and safety of tazarotene 0.1% plus clindamycin 1% gel versus adapalene 0.1% plus clindamycin 1% gel in facial acne vulgaris: a randomized, controlled clinical trial. Clin Drug Investig. 2017;37(11):1083–1091.
- Dhawan SS, Gwazdauskas J. Clindamycin phosphate 1.2%-benzoyl peroxide (5% or 2.5%) plus tazarotene cream 0.1% for the treatment of acne. Cutis. 2013;91(2):99–104.
- Tanghetti E, Dhawan S, Green L, et al. Clinical evidence for the role of a topical anti-inflammatory agent in comedonal acne: findings from a randomized study of dapsone gel 5% in combination with tazarotene cream 0.1% in patients with acne vulgaris. J Drugs Dermatol. 2011;10(7):783–792.
- Epstein EL, Stein Gold L. Safety and efficacy of tazarotene foam for the treatment of acne vulgaris. Clin Cosmet Investig Dermatol. 2013;6:123–125.
- Del Rosso JQ, Tanghetti E. A status report on topical tazarotene in the management of acne vulgaris. J Drugs Dermatol. 2013;12(3):s53–s58.
- Smith JA, Narahari S, Hill D, Feldman SR. Tazarotene foam, 0.1%, for the treatment of acne. Expert Opin Drug Saf. 2016;15(1):99–103.
- Patel MR, Patel RB, Parikh JR, Patel BG. Novel microemulsion-based gel formulation of tazarotene for therapy of acne. Pharm Dev Technol. 2016;21(8):921–932.