
Brent D. Michaels, DO, Dermatology Resident (PGY-2), Valley Hospital Medical Center, Las Vegas, Nevada; James Q. Del Rosso, DO, FAOCD, Dermatology Residency Director, Valley Hospital Medical Center, Las Vegas, Nevada, and Director of Dermatology Research, Mohave Skin & Cancer Clinics, Las Vegas, Nevada;
Narciss Mobini, MD, University of Nevada School of Medicine, Las Vegas, Nevada; Jason R. Michaels, MD, Clinical Instructor (Dermatology), Valley Hospital Medical Center, Las Vegas, Nevada
Abstract
Erythropoietic protoporphyria is considered a rare disease overall, but in children is the most common form of porphyria, and certainly the most common type of erythropoietic porphyria. Despite this fact, erythropoietic protoporphyria is a disease that has been known to evade or at least delay diagnosis, leading to unnecessary suffering by the patient. Given the distress it may cause a patient and his or her family as well as the potential complications of this disease, the importance of maintaining a heightened awareness when presented with a child complaining of photosensitivity cannot be overstated. This case report will review the important clinical indicators, pathogenesis, histology, diagnosis, management, and treatment of this disease, so that affected children will no longer have to play “hide and seek” when diagnosed with this sun-sensitive disease. (J Clin Aesthetic Dermatol. 2010;3(7):44–48.)
There are several types of cutaneous porphyrias that are grouped into one of two categories: hepatic porphyrias and erythropoietic porphyrias. In erythropoietic porphyrias, the excess of porphyrins is mainly found in the red cells.[1] Erythropoietic protoporphyria (EPP) is a type of erythropoietic porphyria and is the most common porphyria found in children.[2] EPP was first clearly defined in 1961 by I.A. Magnus et al[3] and is clinically characterized by photosensitivity to visible light with subsequent physical cutaneous signs in the skin exposed to sun. Photosensitivity with cutaneous lesions usually presents in infancy or early childhood. However, there have been cases of delayed diagnosis resulting from either a late onset of symptoms, patients with only mild symptoms, or simply from a failure to diagnose. As a result, dermatologists must employ a keen awareness for EPP in children with subjective photosensitivity even without associated clinical cutaneous findings.
Case Report
A five-year-old Caucasian girl presented to the emergency room in August with a complaint of swelling of both hands extending to the mid forearms accompanied by diffuse tender purpuric patches on the dorsal surface of the hands. Swelling was noted in the feet, and the patient was found to have swelling of the face with mild purpura confined to the nose. The patient’s cutaneous symptoms were noted to have begun the day before when she was at a lake and exposed to the sun for about five hours. The patient was subsequently admitted with a working diagnosis of angioedema. There were no prior hospital admissions for this condition. However, the patient’s father reported a prior history of “recurrent” episodes of hand and facial swelling that also seemed to occur from prolonged exposure to sunlight and/or heat. The child had been seen by a previous dermatologist and pediatrician and given the diagnosis of photosensitivity. The patient had previously consulted an allergist who recommended biopsy and allergy testing. The allergy workup for medicinal and environmental substances was negative.
On physical exam, non-pitting edema and purpuric patches were noted as above. No jaundice was identified. Apart from one instance of emesis after dexamethasone and diphenhydramine, the review of systems was negative. The patient’s laboratory work, including a complete blood count with manual differential and platelets, a hepatic panel, and urinalysis, was within normal limits. Except for the history of prior edema to sunlight, the past medical history was listed as “completely benign.”
Both a dermatologist and pediatric hematologist were consulted with an original working differential diagnosis of angioedema versus urticarial vasculitis. Several other laboratory values were ordered, including C1 esterase inhibitor, complement C4, complement C3, proteinase 3, myeloperoxidase Abs, ANA, anti-DNA DS, ANCA with reflex, anti-SSA, and anti-SSB. All were within normal limits.
A skin biopsy was performed for evaluation. The initial interpretation stated, “within normal limits, no pathological changes seen.” A second review by another pathologist stated, “highly suggestive of urticarial vasculitis, recommending serological tests to be done for complement deficiencies.” The tests were all negative as noted above. The skin biopsy was then reviewed by another dermatopathologist and the final diagnosis of EPP was rendered based on the following subtle histopathological findings: slight thickening of small blood vessel walls secondary to the presence of pale-eosinophilic material that strongly stained positive with PAS stain, extravasation of erythrocytes, but no vascular damage. Accordingly, laboratory investigation for detecting the levels of porphyrins was recommended. A free erythropoietic protoporphyrin level was ordered, but the result was not available before discharge.
The patient was subsequently followed up as an outpatient shortly after discharge by dermatology. In the erythrocyte sample, coproporphyrins were slightly elevated at 6µg/dL (normal value 0–2µg/dL) although the free protoporphyrins were significantly elevated at 454µg/dL (normal value 16–60µg/dL). Total porphyrins were also elevated at 800µg/dL. Fractional protoporphyrin analysis was performed and chromatography revealed the major form of protoporphyrin was free protoporphyrin. The urine porphyrin levels were normal. The laboratory values thus confirmed the diagnosis of EPP. The patient’s cutaneous lesions subsequently improved through simple sun avoidance and the patient continues to be followed by dermatology.
Discussion
This case emphasizes the enigmatic nature of this disease and the difficult nature of diagnosis if EPP is not considered in children with photosensitivity. Also, in histopathological evaluation, the changes can be subtle and easily missed. While the patient had a prior history of recurrent edema to sunlight, a diagnosis was not made by a previous allergist or dermatologist. The patient continued to have bouts of edema with sun exposure. Thus, even when cutaneous symptoms are present, there are instances when EPP evades diagnosis. Complicating this clinical picture are times when the skin features, beyond photosensitivity, have a delayed onset or are minimal to absent, leaving the patient and family with little hope of diagnosis. There are reports of delayed diagnosis for an average of 10 to 20 years, resulting in considerable distress to the patient and family.[4] Also, as prior published reports note, an early diagnosis will help provide the patient and family legitimacy for their suffering that may otherwise be overlooked as hysteria or hypochondria.[4,5] Thus, an understanding of the clinical manifestations of EPP is essential for any dermatologist who treats patients with photosensitivity.
Clinical features. There are essentially two main clinical manifestations of the inherited disorder of EPP: photosensitivity and hepatobiliary disease.[6] Photosensitivity often affects the sun-exposed skin and usually will be more predominate in the warmer months of spring and summer.[5] Although sunlight is the usual trigger, photosensitivity may also be exacerbated by heat exposure and temperature gradients.[7] Sensations of photosensitivity have been described as skin pain, burning, tingling, prickling, itching, and stinging and usually occur shortly after sun exposure.[2] The burning pain sensation may be severe and cause children to lose sleep for nights.[8] According to one published report, children will try to relieve the pain by applying cold water to the skin, which is noted to be both characteristic and diagnostically helpful.[8] Children may only present with a history of screaming or skin pain when exposed to sunlight with no visible lesions.[2] This can lead to the diagnosis of malingering or hypochondria in some patients without clinical symptoms.[5,6]
Visible changes to sunlight can also vary and include edema, painful erythema, petechiae, purpura, and fissuring.[2,7,9] The diffuse edematous lesions have been identified as resembling angioneurotic edema.10 According to a study by Holme et al,[11] visible changes usually are related to the duration of exposure, with minimal exposure tending not to result in any visible lesions. However, characteristic changes of severe burning pain with erythema and edema have been noted to occur within minutes of light exposure.12 On the other hand, skin changes such as blistering, erosions, crusting, vesicles, and bullous lesions are not as typical as they are in other porphyria disease states, but may occasionally occur with prolonged exposure.[9] The distribution of the lesions also provide diagnostic insight. Location of the skin lesions are characteristic of a photodermatitis, with lesions generally occurring on areas exposed to light.[13] It has been found that the most serious lesions on the face occur on the areas exposed to the highest doses of sunlight and the areas that are more shaded typically are not affected.[13]
Non-skin symptoms have also been reported and involved difficulty with sleep, irritability, temperature insensitivity, nausea, headache, and a depressed state.[11] Nail unit changes are also possible with EPP, including photo-onycholysis.[14] If EPP diagnosis is delayed, repeated sun exposure can lead to more chronic skin changes, such as a waxy thickening of the face and knuckles.[2] Purpuric changes have also been noted on the dorsum of the hands and forearms.[9] Additionally, small erosions on the face leading to shallow circular or linear scarring may be present.[9]
In addition to cutaneous photosensitivity changes, hepatobiliary disease is also a potential clinical finding. According to one report, hepatobiliary disease affects about 25 percent of patients.[15] Protoporphyrin is cleared by the hepatobiliary system and has a concentration-dependent hepatotoxic effect, and thus, accumulation in the liver can impair the hepatobiliary system and potentially lead to liver failure.[15] However, the degree of severity of hepatobiliary disease is highly variable. Patients may present with findings ranging from mild changes in liver function to cholelithiasis and hepatic failure.
The incidence of cholelithiasis in EPP has been reported to be as high as 20 percent of patients and can occur at a young age.[14,16] Gallstones typically contain precipitated protoporphyrin. Conversely, hepatic failure is much less common. Incidence rates have been reported anywhere from 2 to 5 percent.[2,16] Clinical findings of acute hepatic failure include accelerated photosensitivity and cholestatis with associated severe upper abdominal pain, jaundice, splenomegaly, and hemolysis.[17] In this situation, early diagnosis of EPP is obviously critical and physicians must consider EPP in patients with such a presentation. Although rare, patients with end-stage liver disease may also develop a neurological syndrome with symptoms of progressive polyneuropathy, swallowing difficulties, and respiratory depression.[14] Mild hypochromic microcytic anemia may also develop in patients with EPP.18 Microcytic anemia is estimated to be found in 20 to 60 percent of patients.
Biochemistry and enzyme deficiency. The group of metabolic diseases known as porphyrias results from an enzymatic deficiency along the heme biosynthetic pathway. There are more than eight enzymes that are involved in the synthesis of heme. The defects along the pathway are mostly partial enzyme deficiencies and not total enzymatic deficiencies, as total enzyme deficiency is not life compatible.[15]
EPP results from a disturbance in the final step of heme synthesis. Namely, EPP results from a partial deficiency in the ferrochelatase (FECH) enzyme, which is the last enzyme along the pathway responsible for the insertion of iron into protoporphyrin IX to form the heme product (Figure 1).[19] FECH enzyme activity is less than 50 percent of its normal level.[15]
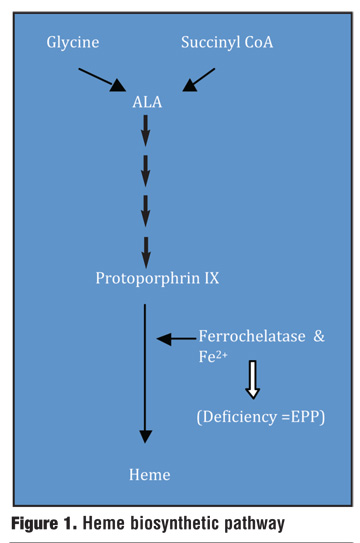{kind=link}
A deficiency in FECH results in an excess of the lipophilic-free protoporphyrin IX. Protoporphyrin IX then accumulates in erythrocytes, liver, plasma, and skin.20 As protoporphyrin is a photoreactive compound, its accumulation in the erythrocytes and plasma of the skin vasculature is what leads to the cutaneous lesions and photosensitivity.[21] The resulting photosensitivity is activated by exposure to ultraviolet (UV) light with a maximum susceptibility at a wavelength of 400nm.[5]
Histology. The histological features of EPP can vary depending on whether acute or chronic changes are present. On light microscopy examination, acute skin reactions will show an intense perivascular and interstitial dermal infiltrate that is largely neutrophilic.[8] Nuclear dust may be present.[8] Additionally, intracellular vacuoles and intercellular edema may also be seen acutely as well as cytolysis of the endothelial cells of superficial blood vessels and degranulated mast cells.[7] As EPP skin lesions progress and the skin becomes chronically damaged, histology will reveal a characteristic amorphous hyaline-like substance that is PAS positive and diastase resistant in the upper dermal capillary wall and upper dermis.[8,13] The capillary vessel walls will also show a marked thickening.[21]
Similar histological changes were also identified in the biopsied lesion in this case. Originally, the dermatopathology findings identified extravascular hemorrhage in the papillary dermis, mild telangiectasia, and the presence of fibrinous thickening of capillary walls, but only noted rare neutrophils in the vessel wall. It was concluded that urticaria and urticarial vasculitis were possibilities, but further serological tests were recommended. On further review by Dr. Mobini (one of the listed authors), an increase in small blood vessels in the papillary dermis was found, most of which showed thickening of their walls secondary to the presence of pale eosinophilic material. There was strong positivity on PAS stain. There was also associated marked extravasation of erythrocytes with minimal inflammatory infiltrate of lymphocytes and scattered erythrocytes.
Although a skin biopsy has been argued to be unnecessary for a diagnosis of EPP,[5] there are instances when it is helpful and can expedite the diagnosis—as it was in this case. Based on the subtle changes and integrating the clinical findings, a diagnosis was made of EPP by dermatopathology that may have otherwise been delayed.
Diagnosis. One of the most important clinical tools for diagnosis is to maintain a high suspicion for EPP when presented with a child complaining of photosensitivity. If suspected, then a progression to laboratory tests is warranted. Diagnostically, the most important clinical indicator of EPP is an elevated free protoporphyrin level in peripheral erythrocytes.[22] Plasma protoporphyrin levels can also be measured. Both elevated free protoporphyrin IX in plasma and erythrocytes is noted as being pathognomonic.[18] Typically, the value of free protoporphyrin is more than five times the normal level.[16] Diagnosis can also be aided by examining for elevated fecal protoporphyrin, as protoporphyrin is eliminated in the feces.[21]
Protoporphyrin is a lipophilic metabolite and thus hydrophobic and not readily soluble in water. As a result, unlike in other porphyrias, the porphyrin levels in urine are normal.[15] Other methods to assist in diagnosis include screening by blood smear fluorescence microscopy to examine for fluorescent erythrocytes, which will show fluorescence of erythrocytes in 5 to 30 percent of cases.[22] For latent cases, an enzymatic test for determination of lymphocyte FECH activity level may also help with confirming diagnosis.[23] As previously mentioned, histological examination can provide further aid in diagnosis.
Once diagnosis is confirmed, additional relevant investigative testing, including liver function tests and appropriate imaging (if hepatobiliary disease is suspected), should be considered.
Treatment and management. Patients with EPP will struggle with photosensitivity their whole lives. Complete sun avoidance to help eliminate visible light exposure is the objective, but is often difficult to achieve. Patients should start by at least modifying sun exposure and implementing lifestyle changes, wearing sun-protective clothing, and applying sun block, such as titanium oxide or zinc oxide. Standard sunscreens that do not block visible light are usually ineffective.[5]
Beta carotene is a treatment modality that has shown effectiveness in reducing photosensitivity in some patients. The clinical effect of oral beta carotene takes about 1 to 3 months, but should be discontinued if there is no significant increase in sunlight tolerance.[22] In one study, beta carotene was discontinued in two-thirds of patients due to such issues as an unacceptable orange discoloration of the skin and body fluids and problems with swallowing the large capsules.[11] It should also be noted that beta carotene is not effective in all patients. Other treatment modalities include cysteine, pyroxidine, vitamin C, antihistamines, terfenadine, and phototherapy using ultraviolet A with psoralen or ultraviolet B lamps. These therapies have also not been shown to be effective in all patients.[5]
If hepatobiliary disease is present, therapy depends on the condition. If cholelithiasis is encountered, surgical management is the likely treatment. For patients who develop liver disease, cholestyramine as well as other porphyrin absorbents, such as charcoal, may be of some benefit to facilitate fecal protoporphyrin excretion.10 If the patient develops acute hepatic failure, liver transplant is the only alternative. Until the transplant is performed, blood transfusions may be of some benefit.[22] It should be noted that while liver transplantation has been found to be life saving in some patients with liver failure, patients may continue to have symptoms of EPP as liver transplantation will not correct the metabolic error.[16] This could result in protoporphyrin damage to the new liver.[10]
Once a patient is diagnosed with EPP, consistent evaluation should be paramount, including performing such tests as a complete blood count, protoporphyrin blood levels, and liver function for any signs of liver damage. If there are any acute changes, such as increasing photosensitivity or physical signs of liver damage, the patient should be followed more closely.
Conclusion
EPP is a disease state that can cause tremendous physical distress to children as well as great psychological distress to parents. This distress may be further compounded by a delayed diagnosis, or, in some cases, a lack of a diagnosis. Moreover, a lack of diagnosis may even become life threatening in cases of severe liver damage. However, much of the distress and sequelae of this disease can be relieved by dermatologists maintaining a keen awareness of EPP in patients who present with photosensitivity irrespective of clinically evident lesions. With this awareness, a more timely diagnosis of EPP can ultimately be made resulting in both appropriate and effective management of the patient.
References
1. Erythropoietic protoporphyria. Br Med J. 1979;2(6203):1456 [No authors listed].
2. Murphy GM. Diagnosis and management of the erythropoietic porphyrias. Dermatol Ther. 2003;16:57–64.
3. Magnus IA, Jarrett A, Prankerd TA, et al. Erythropoietic protoporphyria: a new porphyria syndrome with solar urticaria due to protoporphyrinaemia. Lancet. 1961;2(7200): 448–451.
4. Wahlin S, Floderus Y, Ros A-M, et al. The difficult clinical diagnosis of erythropoietic protoporphyria. Physiol Res. 2006;55(Suppl 2):S155–S157.
5. Lecluse AL, Kuck-Koot VC, van Weelden H, et al. Erythropoietic protoporphyria without skin symptoms—you do not always see what they feel. Eur J Pediatr. 2008;167(6):703–706.
6. Poh-Fitzpartick MB. Erythropoietic protoporphyria. http://emedicine.medscape.com/article/1104061-overview. Accessed on June 9, 2010.
7. Lecha M. Erythropoietic protoporphyria. Photodermatol Photoimmunol Photomed. 2003;19(3):142–146.
8. Todd DJ. Erythropoietic protoporphyria. Br J Dermatol. 1994;131(6):751–766.
9. Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124(1):xvi–xvii.
10. Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135(3):281–292.
11. Holme SA, Anstey AV, Finlay AY, et al. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155(3):574–581.
12. Cox TM. Erythropoietic protoporphyria. J Inherit Metab Dis. 1997;20(2):258–269.
13. Baart de la Faille H, Bijlmer-Iest JC, van Hattum J, et al. Erythropoietic protoporphyria: clinical aspects with emphasis on the skin. Curr Probl Dermatol. 1991;20:123–134.
14. Lecha M, Puy H and Deybach JC. Erythropoietic protoporphyria. Orphanet J Rare Dis. 2009;4:19.
15. Gross U, Hoffmann GF, Doss MO. Erythropoietic and hepatic porphyrias. J Inherit Metab Dis. 2000;23(7): 641–661.
16. Schneider-Yin X, Gouya L, Meier-Weinand A, et al. New insights into the pathogenesis of erythropoietic protoporphyria and their impact on patient care. Eur J Pediatr. 2000;159(10):719–725.
17. Cohen JL, Lim HW. The cutaneous porphyrias. Curr Probl Dermatol. 2000;12(1):35–40.
18. Cox TM, Alexander GJ, Sarkany RP. Protoporphyria. Semin Liver Dis. 1998;18(1):85–93.
19. Herrero C, To-Figueras J, Badenas C, et al. Clinical, biochemical, and genetic study of 11 patients with erythropoietic protoporphyria including one with homozygous disease. Arch Dermatol. 2007;143(9): 1125–1129.
20. Parker M, Corrigall AV, Hift RJ, et al. Molecular characterization of erythropoietic protoporphyria in South Africa. Br J Dermatol. 2008;159(1):182–191.
21. Mather MK, Sau P. Pathological case of the month. Erythropoietic protoporphyria. Arch Pediatr Adolesc Med. 1998;152(6):603–604.
22. Labrousse AL, Salmon-Ehr V, Eschard C, et al. Recurrent painful hand crisis in a four-year-old girl, revealing an erythropoietic protoporphyria. Eur J Dermatol. 1998;8(7): 515–516.
23. Marko PB, Miljkovic J, Gorenjak M, et al. Erythropoietic protoporphyria patients in Slovenia. Acta Dermatovenerol Alp Panonica Adriat. 2007;16(3):99–102, 104.