
Lisa Gruson, MD; Thomas Berk, BA
New York University School of Medicine
Abstract
Hermansky-Pudlak syndrome is an autosomal recessive disorder of lysosomal storage characterized by the triad of occulocutaneous albinism, bleeding diathesis, and pulmonary fibrosis. Sarcoidosis is a disease characterized by the development of noncaseating granulomas, most commonly affecting the lungs. The pathophysiology, histological findings, clinical symptoms, and treatment of the pulmonary manifestations of Hermansky-Pudlak syndrome are distinct from those of sarcoidosis. As patients with occulocutaneous and bleeding manifestations of Hermansky-Pudlak syndrome may also develop pulmonary fibrosis, the authors present this case to illustrate that pulmonary symptoms must be carefully evaluated in those with this syndrome because in this case, the patient developed underlying pulmonary sarcoidosis. To the authors’ knowledge, this is the first documented case of Hermansky-Pudlak syndrome with concomitant pulmonary sarcoidosis. (J Clin Aesthetic Dermatol. 2009;2(10):41–44.)
When treating patients with Hermansky-Pudlak syndrome (HPS), healthcare providers may consider respiratory symptoms a manifestation of HPS-associated pulmonary fibrosis. This form of pulmonary fibrosis has a progressive course, is difficult to treat, and portends a poor prognosis with a shortened life expectancy. Conversely, pulmonary sarcoidosis responds to corticosteroids and, depending on the stage of disease, often has a significantly better prognosis than HPS-associated lung disease. The authors report an unusual case of a patient who was previously diagnosed with HPS-associated pulmonary fibrosis and on further evaluation was found to have pulmonary sarcoidosis. This case illustrates the importance of understanding the systemic manifestations associated with inherited HPS and the need for appropriate consultations and workup of new symptoms in such patients. Additionally, in patients with HPS, a complete pulmonary evaluation may be indicated to determine the exact etiology of the symptoms.
Case Report
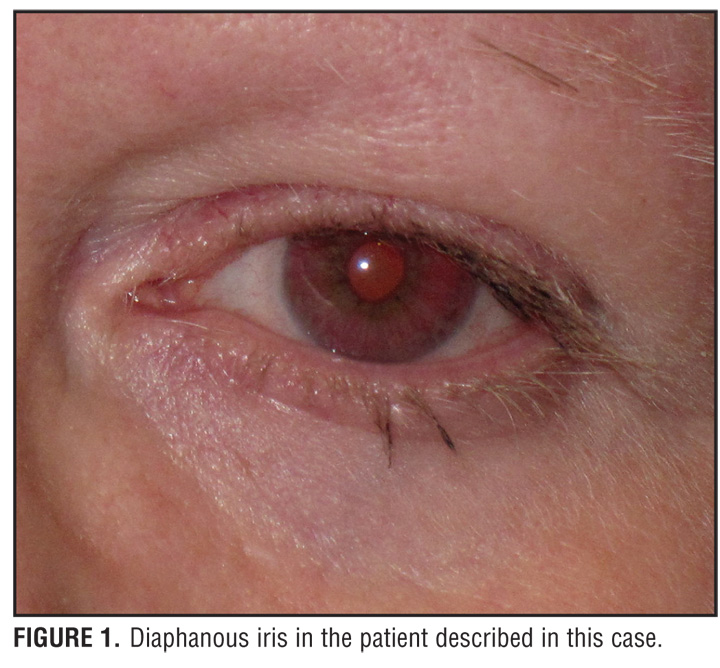
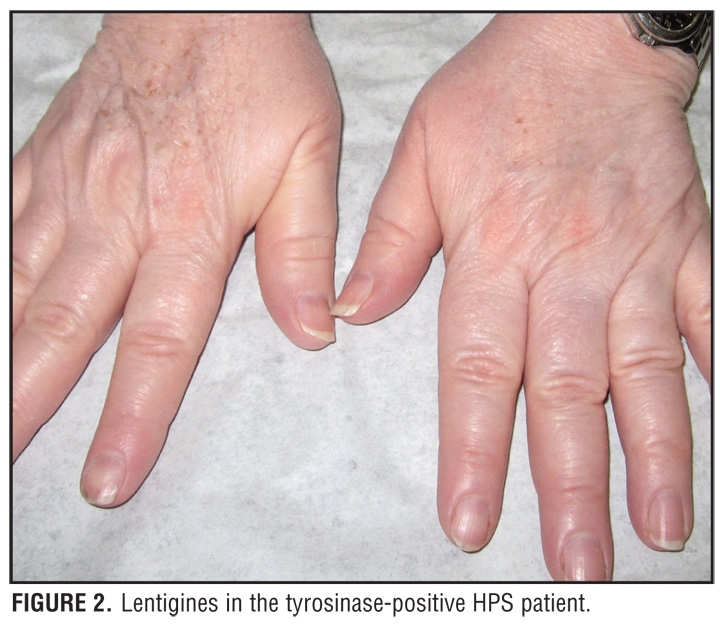
A 59-year-old Puerto Rican woman with HPS who was recently diagnosed with pulmonary sarcoidosis had experienced lifelong occulocutaneous albinism (OCA) and a bleeding diathesis, but was undiagnosed until she read a story in the local newspaper about a family with HPS (Figure 1 and Figure 2). She was subsequently diagnosed with HPS through laboratory studies revealing an absence of platelet-dense bodies on electron microscopy, indicating a platelet storage deficiency. This, in combination with her history of OCA and a prolonged bleeding time of 18.5 minutes, was diagnostic of HPS.
{kind=link}
{kind=link}
The patient remained in good health without other significant laboratory abnormalities until 2001, when she began experiencing dyspnea on exertion. Her symptoms were first attributed to pulmonary fibrosis secondary to HPS. However, a diagnostic pulmonary workup, including a high-resolution computed tomography (CT) scan as well as multiple pulmonary function tests, yielded results inconsistent with pulmonary fibrosis. Her symptoms continued to progress and further imaging and pulmonary function testing did not reveal HPS-associated pulmonary fibrosis. The constitutional symptoms continued until 2007, when she was noted on a follow-up, high-resolution, chest CT to have bilateral hilar lymphadenopathy and diffuse pulmonary nodules. A subsequent lung biopsy of the right upper lobe demonstrated the presence of noncaseating granulomata consistent with a diagnosis of pulmonary sarcoidosis. A tapering course of prednisone resulted in resolution of her current symptoms, which resolved over the next 2 to 3 months. The patient did not develop any cutaneous symptoms of sarcoidosis and her disease was limited to her lungs.
Discussion
HPS comprises a rare group of autosomal recessive disorders characterized by the triad of OCA, bleeding diathesis, and, in many cases, the accumulation of ceroid lipofuscin, resulting in pulmonary, gastrointestinal, and cardiac manifestations. HPS, associated with defects in seven genes, is caused by the impairment of intracellular trafficking of three membrane-bound organelles—melanosomes, platelet-dense bodies, and lysosomes.[1] This condition may well be the most common, single, genetic disorder in Puerto Rico, with a prevalence of 1 in 1,800, and an estimated carrier rate of 1 in 22.[2,3] The impaired intracellular trafficking of melanin in the melanocytes of the skin and retina are thought to be the cause of the OCA; whereas, impaired formation of platelet-dense bodies leads to the bleeding dyscrasia.
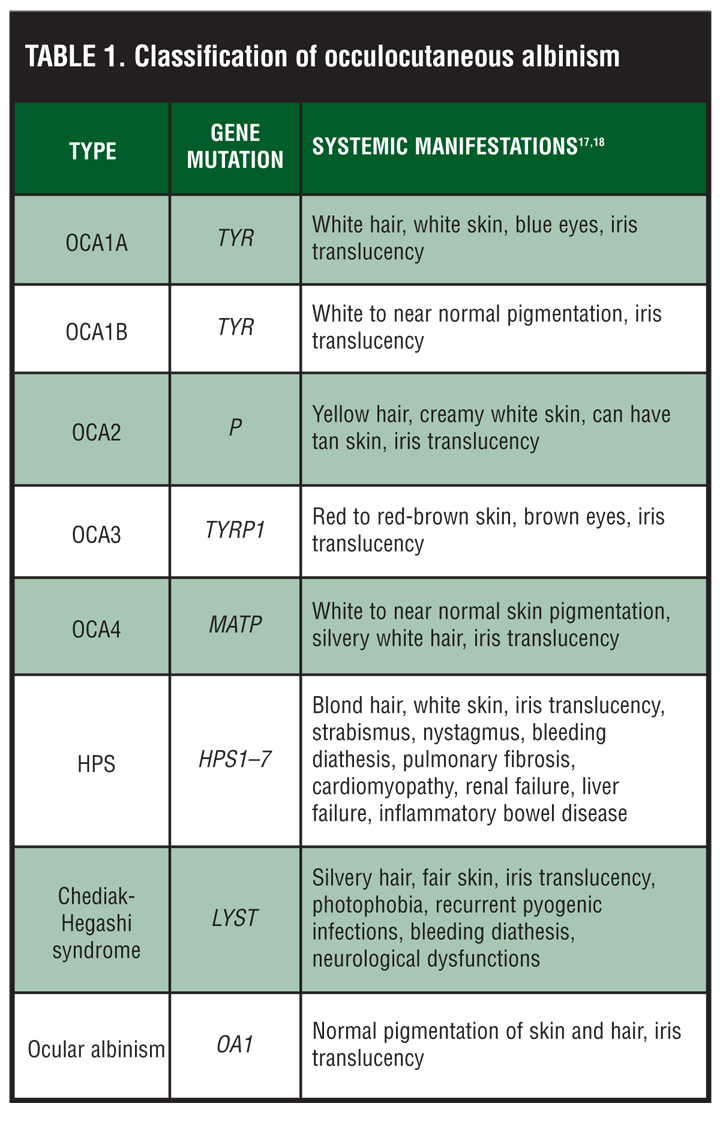
Patients with HPS have the tyrosinase-positive form of OCA. For this reason, the skin, hair, and eyes may have some pigment present. Hair color may range from cream to red-brown in color. In addition, pigmented nevi and lentigines may also be present on the skin, especially from age 10 onward.[2] Ophthalmological findings include nystagmus, photophobia, decreased visual acuity, and diaphanous irides (Table 1).[2]
{kind=link}
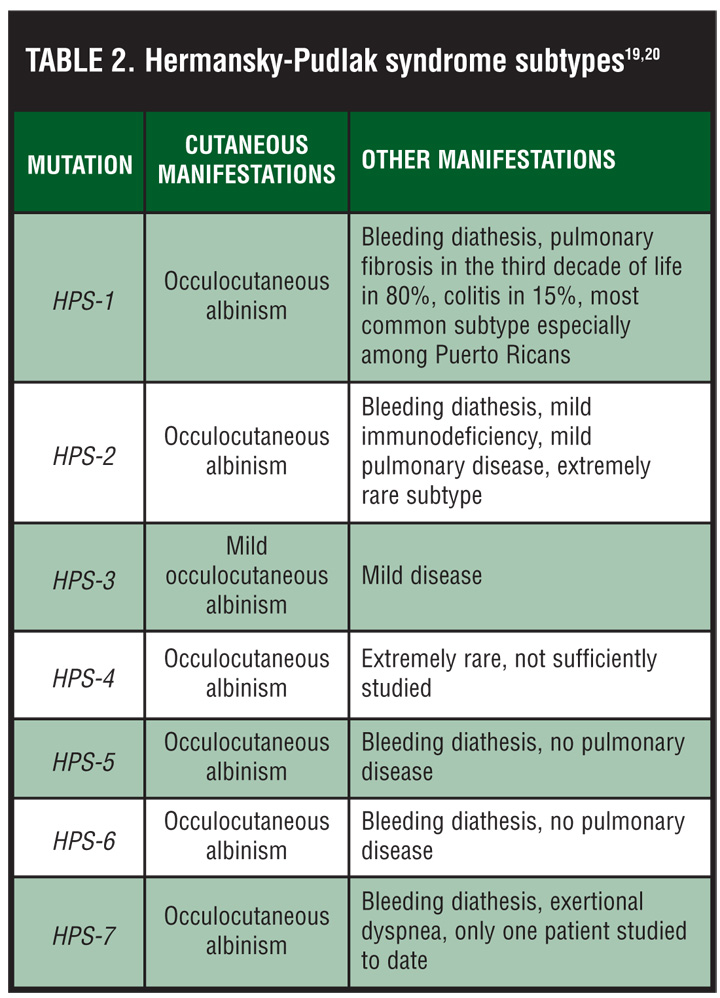
The pulmonary, cardiac, and gastrointestinal manifestations of HPS are due to ceroid lipofuscin accumulation within lysosomes throughout the body. This may lead to pulmonary fibrosis, cardiomyopathy, and granulomatous colitis, respectively. The lifespan of patients with these severe disease subtypes usually does not exceed 40 to 50 years.[4–6] The pulmonary fibrosis associated with HPS occurs in 80 percent of patients with the most common mutation, a 16-bp duplication in exon 15 of the HPS-1 gene, leading to premature mortality in approximately 50 percent of cases (Table 2).[7,8] The manifestation of pulmonary fibrosis in HPS is disappointingly difficult to treat as immunosuppressives and anti-inflammatories have little impact on ceroid deposition. However, the anti-inflammatory and antifibrotic pirfenidone has shown efficacy in treating chemotherapy-induced pulmonary fibrosis in mice,[9] and may have some efficacy in delaying the progression of HPS-associated fibrosis as well.[10]
{kind=link}
Currently, the protein structure of the most common genetic mutation in HPS—the HPS-1 gene, a 700 amino acid protein associated with chromosome 10q2—is being investigated11 (Berk T, personal communication, 2007–2009, New York, NY), and the protein structure of a splice site mutation in the HPS-1 gene is being ascertained. This knowledge will hopefully lead to a better understanding of the methods of intracellular trafficking in general, and for this challenging disease specifically.[12]
Patients with HPS are predisposed to numerous medical conditions and therefore need to be followed regularly as outpatients by multiple specialists. Their deficiency in melanin production leads to an increased risk of skin cancers, requiring regular dermatological examinations. HPS patients should be counseled regarding the importance of sun protection and being self vigilant. They should also be taught the techniques to perform skin self examinations. HPS patients should also be aware of their bleeding diathesis and will need consultation with a hematologist regarding proper care if they do begin to bleed. Ophthalmology, pulmonary, gastroenterology, and cardiology referrals may also be indicated.
This is in distinction from the very different disease process of sarcoidosis. Sarcoidosis is a multisystem disease characterized by the presence of noncaseating epithelioid granulomas thought to be a pathological response to foreign material. A diagnosis of sarcoidosis is established through thorough clinical examination, radiographic findings, and the histopathological diagnosis of noncaseating granulomas. Sarcoidosis can affect many organ systems, and approximately 25 percent of affected individuals will have skin involvement.[13] Cutaneous lesions in sarcoidosis may be either specific lesions that contain granulomas, such as papules, nodules, plaques, or lupus pernio; or nonspecific reactive processes including erythema nodosum.[13] Pulmonary involvement occurs in more than 90 percent of cases and most commonly presents as a cough with dyspnea.[14] Radiographically, pulmonary sarcoidosis involves four stages: bilateral hilar lymphadenopathy (BLH) without pulmonary infiltrates, BLH with infiltrates, infiltrates without BLH, and extensive fibrosis with possible formation of bullae. On pulmonary function testing, 40 percent of individuals with stages 2 to 4 pulmonary sarcoidosis show a restrictive pattern[15] with airflow obstruction, as evidenced by decreased FEV-1 and expiratory flow rates, which are seen in up to two-thirds of such cases.[16]
Treatment of sarcoidosis is based on the organ systems involved and the resulting symptoms. As this disease may sometimes spontaneously resolve, initial observation without therapy may be prudent. With regard to pulmonary involvement, treatment is indicated starting when a patient has symptomatic or progressive stage 2 disease. Corticosteroid therapy has been shown to induce remission in 50 to 60 percent of patients with moderate (stage 2 or 3) sarcoidosis.[17]
Many dermatologists treat patients with various forms of OCA, and the association of HPS with pulmonary fibrosis is well known. In addition, although our patient did not have any cutaneous manifestations of sarcoidosis, if present, these could have been used to guide the diagnostic process. Although, after a review of the literature, there appears to be no increased likelihood of HPS patients developing sarcoidosis, this report highlights the importance of the treating dermatologist considering the systemic manifestations that may be associated with albinism and to advise patients to receive the appropriate consultations and workup. Even patients with a diagnosed condition, such as HPS, should undergo a complete workup if new symptoms arise. The advantage to confirming a diagnosis of sarcoidosis is the ability to treat pulmonary symptoms with corticosteroid therapy. Occulotoxic drugs and anticoagulants should be avoided or used with caution in patients with HPS.
In the case presented, the thorough pulmonary evaluation pointed to a diagnosis of an unrelated syndrome, sarcoidosis. Although there is no effective treatment for HPS pulmonary fibrosis, corticosteroid therapy is highly efficacious in the treatment of sarcoidosis.
References
1. Huizing M, Anikster Y, Gahl WA. Hermansky-Pudlak syndrome and Chediak-Higashi syndrome: disorders of vesicle formation and trafficking. Thromb Haemost. 2001;86(1):233–245.
2. Witkop CJ, Nuñez Babcock M, Rao GH, et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol Asoc Med P R. 1990;82(8):333–339.
3. Anikster Y, Huizing M, White J, et al. Mutation of a new gene causes a unique form of Hermansky-Pudlak syndrome in a genetic isolate of central Puerto Rico. Nature Genetics. 2001;28:376–380.
4. Boissy RE, Richmond B, Huizing M, et al. Melanocyte-specific proteins are aberrantly trafficked in melanocytes of Hermansky-Pudlak syndrome-type 3. Am J Pathol. 2005;166(1):231–240.
5. Hermos CR, Huizing M, Kaiser-Kupfer MI, Gahl WA. Hermansky-Pudlak syndrome type 1: gene organization, novel mutations, and clinical-molecular review of non-Puerto Rican cases. Hum Mutat. 2002;20(6):482.
6. Harmon KR, Witkop CJ, White JG, et al. Pathogenesis of pulmonary fibrosis: platelet-derived growth factor precedes structural alterations in the Hermansky-Pudlak syndrome. J Lab Clin Med. 1994;123(4):617–627.
7. Shotelersuk V, Dell’Angelica EC, Hartnell L, Bonifacino JS, Gahl WA. A new variant of Hermansky-Pudlak syndrome due to mutations in a gene responsible for vesicle formation. Am J Med. 2000;108(5):423–427.
8. Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky-Pudlak syndrome type 4 (HPS-4): clinical and molecular characteristics. Hum Genet. 2003;113(1):10–17.
9. Nicod LP. Pirfenidone in idiopathic pulmonary fibrosis. Lancet. 1999;354(9175):268–269.
10. Gahl WA, Brantly M, Troendle J, et al. Effect of pirfenidone on the pulmonary fibrosis of Hermansky-Pudlak syndrome. Mol Genet Metab. 2002;76(3):234–242.
11. Oh J, Bailin T, Fukai K, et al. Positional cloning of a gene for Hermansky-Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet. 1996;14(3):300–306.
12. Oh J, Ho L, Ala-Mello S, Amato D, et al. Mutation analysis of patients with Hermansky-Pudlak syndrome: a frameshift hot spot in the HPS gene and apparent locus heterogeneity. Am J Hum Genet. 1998;62(3):593–598.
13. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol. 2001;44(5):725–743.
14. Roberts SD, Mirowski GW, Wilkes D, Teague SD, Knox KS. Sarcoidosis. Part I: pulmonary manifestations. J Am Acad Dermatol. 2004;51(3):448–451.
15. Alhamad EH, Lynch JP 3rd, Martinez FJ. Pulmonary function tests in interstitial lung disease: what role do they have? Clin Chest Med. 2001;22(4):715–750, ix.
16. Sharma OP, Johnson R. Airway obstruction in sarcoidosis. A study of 123 nonsmoking black American patients with sarcoidosis. Chest. 1988;94(2):343–346.
17. Newman LS, Rose CS, Maier LA, Sarcoidosis. N Engl J Med. 1997;336(17):1224–1234.
18. Lyle WM, Sangster JO, Williams TD. Albinism: an update and review of the literature. J Am Optom Assoc. 1997;68(10):623–645.
19. Spitz JL. Genodermatoses. 2nd ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2005.
20. Morgenthau AS, Padilla ML. Spectrum of fibrosing diffuse parenchymal lung disease. Mt Sinai J Med. 2009;76(1):2–23.